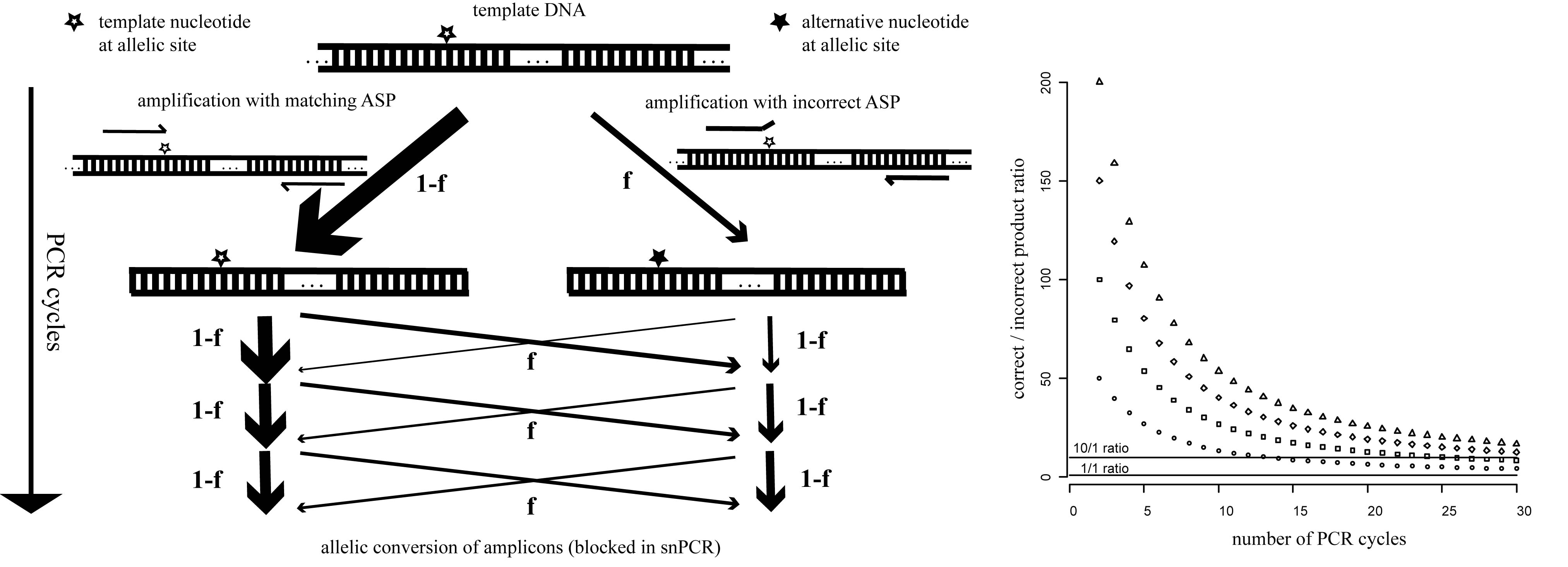
Allele-specific PCR (AS-PCR) is an inexpensive and fast method to discriminate genotypes based on single nucleotide differences. It relies on the requirement of Taq polymerase for valid base-pairing between the template and the 3’-most nucleotide of allele-specific primers (ASPs). In its simplest form, an AS-PCR combines three primers in one reaction: two ASPs, each of them matching one of the two allelic nucleotides at its 3’-most position, and a common reverse primer. For the single-tube reaction, at least one of the ASPs is tagged to distinguish the alternative products, e.g., a size tag of 5’ non-specific nucleotides allows for separation by gel electrophoresis. Unfortunately, the method requires extensive optimization of conditions to obtain sufficient specificity for unambiguous genotyping, which has hindered its wide-spread use in genotyping SNP alleles and is preventing its use in SNP mapping.
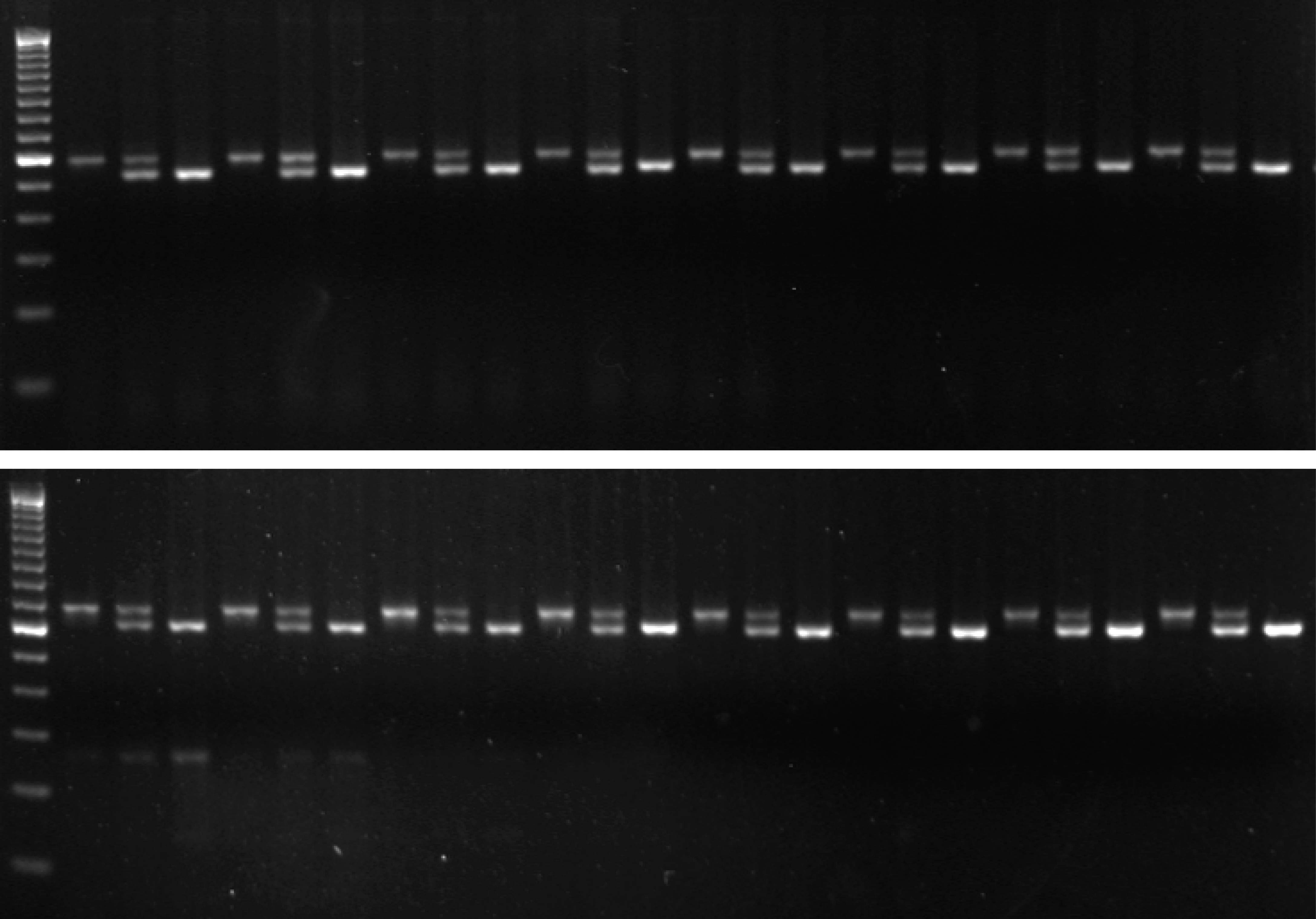
We realized that the amplification process limits specificity of single-tube AS-PCR because relatively rare events of mispriming generate allele-switched amplicons that are propagated during subsequent cycles (Fig. 1). We found that incorporating additional mismatches into the ASPs that make them different from both the template and one another at the third-last position prevents this problem because a single round of amplification now results in amplicons with a perfect match to one ASP, but two mismatches to the other (which is not achieved by introducing a common mismatch into the ASPs as sometimes suggested). Thus, mispriming is effectively limited to the original template and the inherent specificity of the polymerase for the matching primer is preserved over any number of cycles. We routinely use this improved snPCR (for PCR with single nucleotide specificity) in the Alcedo lab to follow genotypes during crosses and found it to be just as robust and reliable as standard PCR (two examples are given in Fig. 2). snPCR works well with standard worm lysates and optimization of conditions is usually not required. For separation of the alternative products we use size tags of 25-30 non-specific nucleotides on one of the two ASPs and 3% agarose (Nusieve 3:1 for the separation of nucleic acids < 1kb) gels. To achieve good separation we design the primers to amplify fragments of < 500 bp. We have also successfully used Cy5/Cy3-labeled ASP pairs to distinguish products based on fluorescence rather than size and read out genotypes from standard gels with a fluorescence scanner. Avoiding interference between the size tags and overcoming the product size limitation this approach should enable the multiplexing of snPCRs and should make the method useful also for SNP mapping. Since relative allele frequencies can be determined with snPCR it should be compatible with the bulked segregant analysis described by the Plasterk group (Wicks et al., 2001) and represent a convenient and fast alternative to allele-specific restriction digests.
During the preparation of this article, we found that the principle behind our snPCR has recently been described elsewhere (Gaudet et al., 2009). The authors state that any mismatches within the last 5 nucleotides of the ASPs - even if they differ in position between the two ASPs - can be chosen, provided they result in two versus zero mismatches after the first round of amplification. This finding is in good agreement with our mechanistic explanation and, if true, makes the design of the ASPs quite flexible.
Figures
References
Gaudet M, Fara AG, Beritognolo I, and Sabatti M. (2009). Allele-specific PCR in SNP genotyping. Methods Mol. Biol. 578, 415-424. 
Wicks SR, Yeh RT, Gish WR, Waterston RH, and Plasterk RH. (2001). Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 28, 160-164.