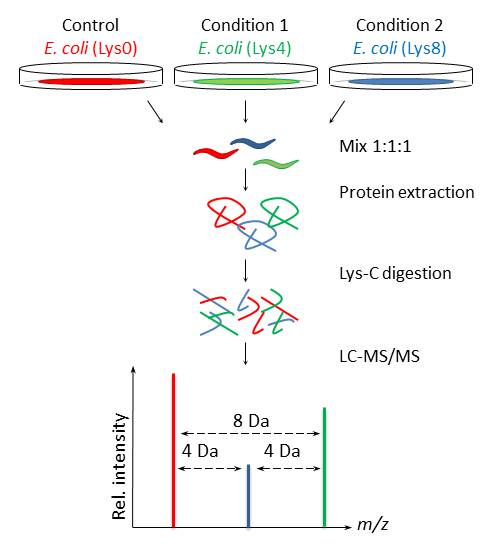
Throughout the biological sciences there is an increasing interest for proteomics to understand cellular processes in response to various treatments. Mass spectrometry has provided the predominant technical platform for such analysis by offering fast and relatively automated large-scale identification and quantification of several thousands of proteins. Besides label-free quantification, several quantification strategies have been developed that are based on stable isotopes having the same physicochemical properties but different masses. These include metabolic labeling, where stable isotopes are incorporated in vivo, which allows samples for comparison to become mixed prior to sample preparation to avoid introduction of quantitative bias. Metabolic labeling with 15N has been used for several studies in C. elegans, including those described by Krijgsveld et al. (2003). In 2002, stable isotope labeling by amino acids in cell culture (SILAC) (Ong et al., 2002; Jiang and English, 2002) was introduced and shown to provide a more precise quantification because the number of labels per peptide is defined, improving data analysis by reducing the complexity of the mass spectra. SILAC is increasingly being used for studies in multi-cellular organisms, and recently we and others applied SILAC to C. elegans (Fredens et al., 2011; Larance et al., 2011).
By a simple procedure we have shown complete incorporation of isotope labeled lysine into C. elegans by feeding the nematode on prelabeled lysine auxotroph E. coli for one generation. Furthermore, we have rendered this E. coli to allow efficient gene knockdown by RNAi in C. elegans. Traditionally, SILAC is based on labeling with lysine and arginine, which provides one label per peptide after digestion with trypsin. However, in C. elegans such quantification is troubled by arginine to proline conversion, so we employed only labeled lysine and used lysyl endopeptidase (Lys-C) for subsequent digestion. Even though the resulting peptides are longer than tryptic peptides and less suited for mass spectrometry, we were able to quantify a vast number of proteins due to intensive peptide fractionation. Larance et al. (2011) included arginine labeling of C. elegans and prevented conversion by RNAi-mediated knockdown of ornithine transaminase, orn-1. However, knockdown of orn-1 may have unwanted effects in, e.g., metabolic or developmental studies, and RNAi against an additional gene may reduce knockdown efficiency (Kamath et al., 2001).
Our present setup allows unbiased comparison of up to three populations of C. elegans to study proteomic changes upon treatment, mutations, or gene knockdown (Figure 1). Furthermore, in combination with enrichment of posttranslational modifications signaling cascades can be studied and we are confident that this approach will facilitate characterization of protein functions in C. elegans.
Figures
References
Fredens J, Engholm-Keller K, Giessing A, Pultz D, Larsen MR, Hojrup P, Moller-Jensen J, and Faergeman NJ. (2011). Quantitative proteomics by amino acid labeling in C. elegans. Nat. Methods 8, 845-847. 
Jiang H, and English AM. (2002). Quantitative analysis of the yeast proteome by incorporation of isotopically labeled leucine. J. Proteome Res. 1, 345-350.
Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, and Ahringer J. (2001). Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2, RESEARCH0002
Krijgsveld J, Ketting RF, Mahmoudi T, Johansen J, Artal-Sanz M, Verrijzer CP, Plasterk RH, and Heck AJ. (2003). Metabolic labeling of C. elegans and D. melanogaster for quantitative proteomics. Nat. Biotechnol. 21, 927-931.
Larance M, Bailly AP, Pourkarimi E, Hay RT, Buchanan G, Coulthurst S, Xirodimas DP, Gartner A, and Lamond AI. (2011). Stable-isotope labeling with amino acids in nematodes. Nat. Methods 8, 849-851.
Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 1, 376-386.