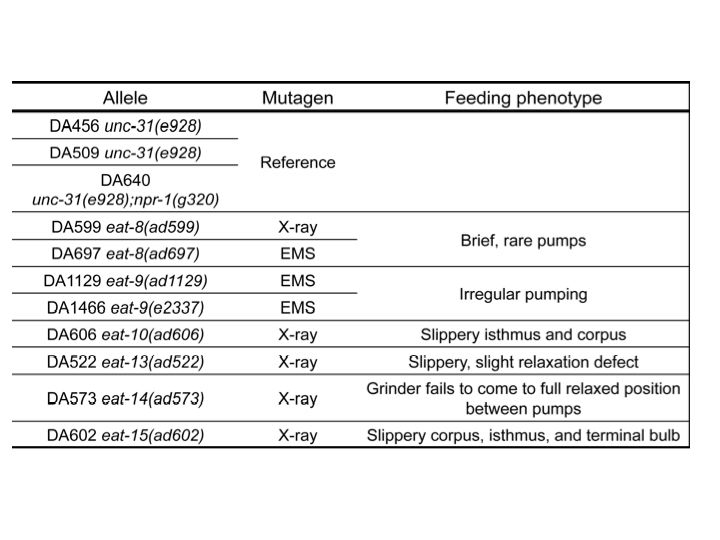
eat mutants, which have feeding defective phenotypes, were isolated many years ago (Avery, 1993). However, eight eat mutants (representing six genes) are not known molecularly (Table 1). To find out these genes, we used whole-genome sequencing (Sarin et al., 2008). Because we had approximate locations from classical genetic mapping, we compared reference and mutant whole-genome sequencing results in the regions to which each gene was known to map.
eat-8(ad599) had deletions and eat-8(ad697) had a missense mutation in Y82E9BR.16. This gene encodes a transporter, a member of the solute carrier family 22 superfamily. Mutations affecting similar proteins are associated with increased risk for Crohn’s disease in Homo sapiens (Newman et al., 2005). We are cloning Y82E9BR.16 to verify its identity as eat-8.
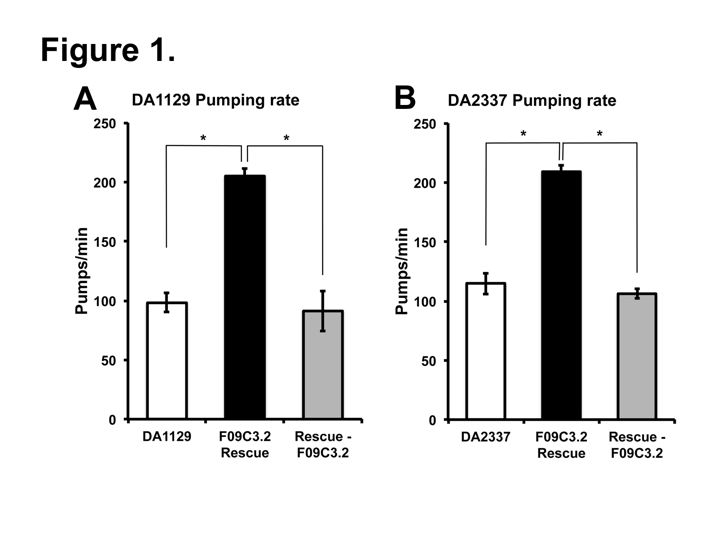
Two independent eat-9 mutants, eat-9(ad1129) and eat-9(e2337), had nonsense mutations at different locations in the coding region of F09C3.2. The eat-9 mutant phenotype was rescued by introducing wild type F09C3.2. When rescued mutants lost wild type F09C3.2, the mutant phenotype was recovered (Fig 1). F09C3.2 encodes a member of the haloacid dehydrogenase (HAD) superfamily (Seifried et al., 2012). We are studying what kind of HAD are related to the irregular pumping phenotype of eat-9.
eat-10 had a frame shift mutation in grld-1, orthologous to human RBM15 and Drosophila NITO, which affects glutamate receptor expression and extends lifespan about 10% by RNAi (Wang et al., 2010; Samuelson et al., 2007). We are comparing the eat-10 mutant phenotype with the known grld-1 mutant phenotypes.
eat-15(ad602) had a deletion in K10D3.4, which encodes a serine protease inhibitor. We cloned and made transgenic worms with this gene and are planning to measure the pumping rate and growth rate and compare the mutant phenotypes.
eat-13(ad522) had a deletion of over 70 kbp, which removed at least 19 genes from R07B1.5 to tsp-21 on chromosome X. We are planning fosmid and cosmid rescue experiments to identify which genes are related to the mutant phenotype. eat-14(ad573) had a 50 kbp deletion which removed at least eight genes from rsd-3 to F54F7.2 on chromosome X. We will also perform fosmid rescue experiments on this mutant. It is of course possible that the mutant phenotypes depend on multiple genes.
In conclusion, we used whole-genome sequencing method to find candidate mutations in 8 eat mutant strains previously believed to define 6 genes. We need more results to verify some of these candidates. However, these results, together with the identification eat‑17 as F01G12.6 and T24D11.1 (see Straud and Avery, this issue WBG) are likely to complete the molecular identification of all the eat genes.
Figures
References
Avery L. (1993) The genetics of feeding in Caenorhabditis elegans. Genetics 133, 897-917. 
Newman B, Gu X, Wintle R, Cescon D, Yazdanpanah M, Liu X, Peltekova V, van Oene M, Amos CI, Siminovitch KA. (2005) A risk haplotype in the Solute Carrier Family 22A4/22A5 gene cluster influences phenotypic expression of Crohn’s disease. Gastroenterology 128, 260–269.
Samuelson AV, Klimczak RR, Thompson DB, Carr CE, Ruvkun G. (2007) Identification of Caenorhabditis elegans genes regulating longevity using enhanced RNAi-sensitive strains. Cold Spring Harb Symp Quant Biol 72, 489–497.
Sarin S, Prabhu S, O'Meara MM, Pe'er I, Hobert O. (2008) Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods 5, 865–867.
Seifried A, Schultz J, Gohla A. (2012) Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J, 549-571.
Wang GJ, Kang L, Kim JE, Maro GS, Xu XZS, Shen K. (2010) GRLD-1 regulates cell-wide abundance of glutamate receptor through post-transcriptional regulation. Nat Neurosci 13, 1489–1495.