Our lab has previously developed a combination of single-step SNP mapping and whole genome sequencing (WGS) methods to pinpoint phenotype-causing sequence variants (Doitsidou et al., 2010). The mapping part of the single-step procedure is essential to separate the phenotype-causing, mutagen-induced sequence variant from background sequence variants. WGS data can be analyzed with CloudMap, a free, cloud-based software analysis tool that we recently developed (Minevich et al., 2012). We have previously provided proof-of-concept studies for the use of CloudMap, and we report here our experience with using CloudMap on a routine basis.
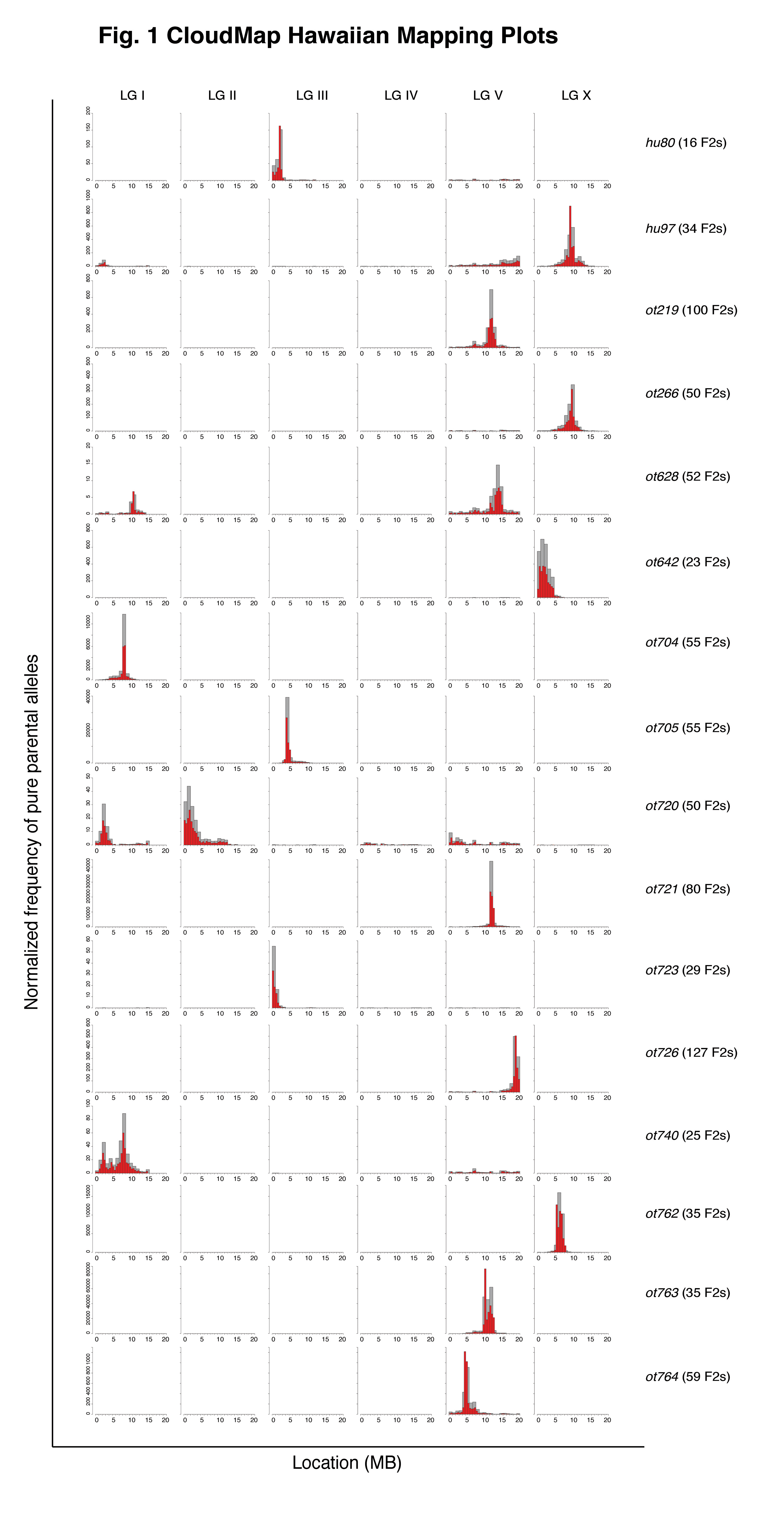
We have found over the last two years that the SNP/WGS strategy and ensuing CloudMap-based data analysis can reproducibly and accurately pinpoint phenotype-causing mutations to mapping regions as small as 0.5 Mb and that the method can work with very few recombinants. Fig. 1 shows the mapping plots from a selection of 16 EMS mutagenized strains from 8 different EMS-induced screens for loss of neuronal identity (thanks to many members of the Hobert lab for providing the mapping data). The tallest histograms indicate the regions containing the largest amount of normalized pure parental alleles following an outcross to the Hawaiian CB4856 mapping strain. For 14 of the causal variants in 13 strains (ot628 is a confirmed double mutant) we have either cloned the causal variant or have strong evidence for its identity and are in the process of confirming. For 8/14 of the causal variants, CloudMap correctly identified the 0.5 Mb where the causal variant resides. In the 6 cases where the 0.5 Mb bins did not correctly identify the causal variant, CloudMap was off by an average of 0.54 Mb.
The accuracy of CloudMap in identifying mapping intervals depends on strict adherence to the Hawaiian SNP mapping protocol and also on the number of F2 recombinants that are pooled for sequencing. In addition, Mendelian ratios should be calculated to identify whether the causal variant resides at one locus or many and if it is dominant or recessive. We also recommend thawing an ancestral Hawaiian strain once a year to minimize variants introduced by genetic drift and to maximize mapping accuracy.
In a number of cases where the Hawaiian mapping protocol was strictly followed, CloudMap was able to identify the correct 0.5 Mb mapping interval with very few recombinants. For example, both hu80 and hu97 were correctly mapped to a 0.5 Mb mapping interval with as few as 16 and 34 F2 recombinants respectively.
We consistently noted that the previously described incompatibility region between Bristol and Hawaian strains on the left arm of LG I (Seidel et al., 2008) is largely not visible relative to the peak of the causal variant due to the normalization procedure we use to compute the frequency of pure parental alleles. In cases where the causal variant is also on LG I (ot740, the taller peak contains the causal variant), CloudMap was able to separate the incompatibility peak from the causal variant peak.
Transgenes contained in the strain background and required to observe a mutant phenotype can generate a mapping signal if the transgene is re-homozygosed in the F2 generation. We find that this mapping signal is virtually eliminated if as few as 50% of the picked F2 recombinants are heterozygous for the transgene.
CloudMap is available via the Galaxy web platform, requires no installation when run on the cloud, and can also be run locally or via the Amazon Elastic Compute Cloud (EC2) service (http://usegalaxy.org/cloudmap and http://hobertlab.org/cloudmap).
Figures
References
Doitsidou M, Poole RJ, Sarin S, Bigelow H, and Hobert O. (2010). C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 5, e15435. 
Minevich G, Park DS, Blankenberg D, Poole RJ, and Hobert O. (2012). CloudMap: A cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269.
Seidel HS, Rockman MV, and Kruglyak L. (2008). Widespread genetic incompatibility in C. elegans maintained by balancing selection. Science 319, 589–594.