Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Nematodes are the most abundant type of animal on earth, and live in hot springs, polar ice, soil, fresh and salt water, and as parasites of plants, vertebrates, insects, and other nematodes. This extraordinary ability to adapt, which hints at an underlying genetic plasticity, has long fascinated biologists. The fully sequenced genomes of Caenorhabditis elegans and Caenorhabditis briggsae, and ongoing sequencing projects for eight other nematodes, provide an exciting opportunity to investigate the genomic changes that have enabled nematodes to invade many different habitats. Analyses of the C. elegans and C. briggsae genomes suggest that these include major changes in gene content; as well as in chromosome number, structure and size. Here I discuss how the data set of ten genomes will be ideal for tackling questions about nematode evolution, as well as questions relevant to all eukaryotes.
In terms of the numbers of individuals, nematodes are the most abundant type of animal on earth (Platt, 1994). So far 25,000 species have been classified, and there could be 100 million species (Blaxter, 2003; Lambshead, 1993). This abundance results from their ability to adapt, as well as their small size, resistant cuticle, and simple body plan. Small changes to their body plan have allowed invasion of many different habitats. Nematodes live in hot springs, polar ice, soil, fresh and salt water, and as parasites of plants, vertebrates, insects, and other nematodes (Andrássy and Zombori, 1976). This evolutionary plasticity, which hints at an underlying genetic plasticity, has long fascinated biologists. In 1965, the German zoologist Alfred Kaestner wrote "our knowledge concerning the evolution of nematodes is next to nothing." Happily, with the genome sequences of the nematodes Caenorhabditis elegans and C. briggsae in hand, and those of C. remanei, C. japonica, C. sp. PB2801, Pristionchus pacificus, Haemonchus contortus, Meloidogyne hapla, Brugia malayi, and Trichinella spiralis soon to follow, our knowledge is now growing fast. This data set of ten genomes will be ideal for tackling questions about nematode evolution, but can also be studied to address questions relevant to all eukaryotes.
In this chapter I will discuss some of the major evolutionary forces that have shaped nematode genomes: how selection seems to have acted to preserve operons and clusters of co-expressed genes (see 3.4 and 3.5); how the involvement of the X and Y chromosomes in sex determination may have affected the evolution of their sequence and structure (see 3.6); how different reproductive strategies may have affected the evolution of chromosome number, structure, and sequence (see 3.1, 3.4 and 3.7); and how a large number of novel genes have arisen and acquired new functions, probably enabling adaptation to new environmental niches, such as parasitism (see 4.3, 4.4 and 4.5).
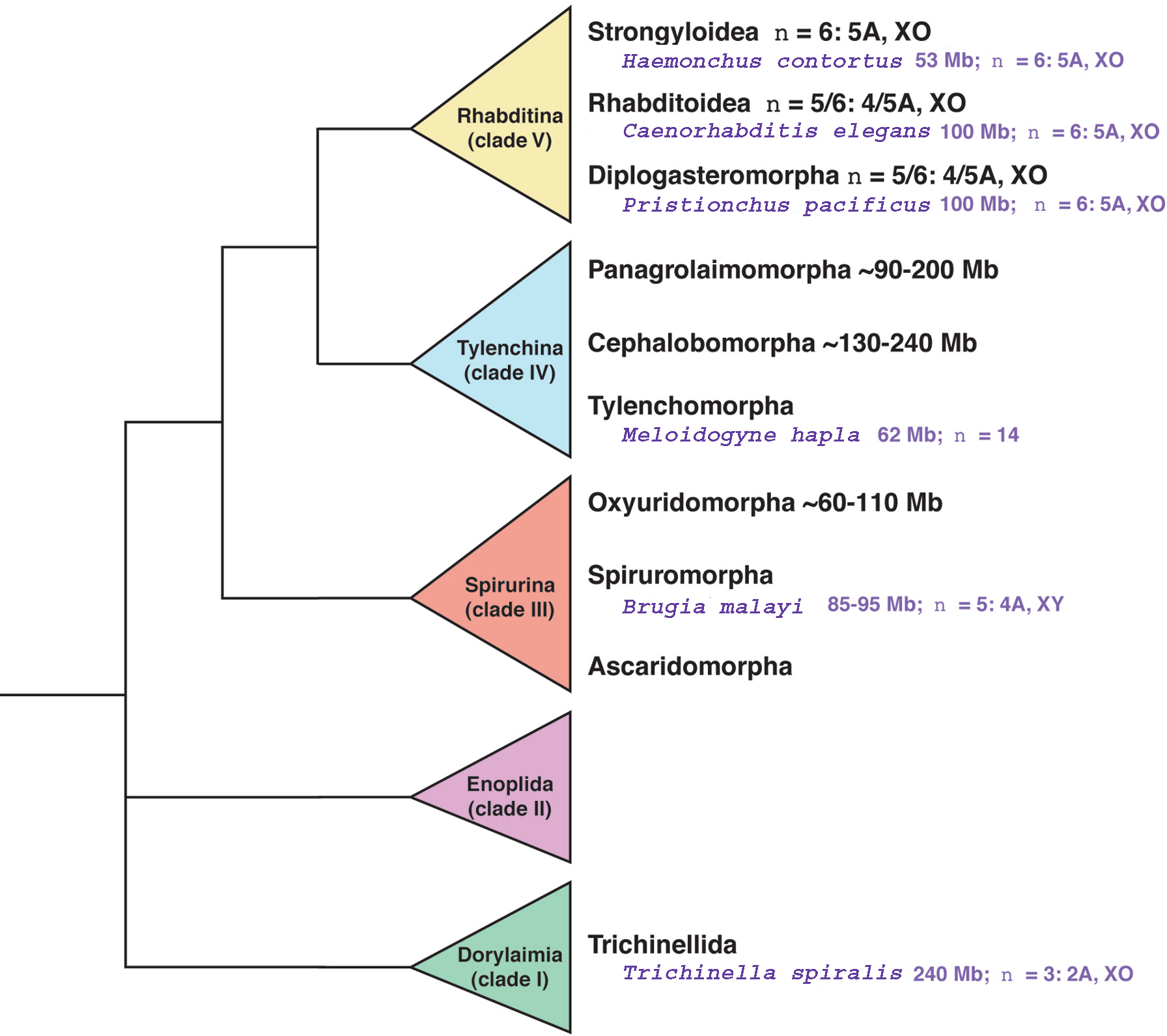
Most nematodes have genomes ranging from 50–250 Mb (Leroy et al., 2003; Figure 1). Among the nematodes being sequenced, sizes vary from 53 Mb for Haemonchus contortus (Leroy et al., 2003) to 240 Mb for Trichinella spiralis (Hammond and Bianco, 1992). A few nematodes even have genomes as large as those of mammals, such as the ~2100 Mb genome of Parascaris univalens (Niedermaier and Moritz, 2000). Other nematode genomes are tiny, such as the ~30 Mb Bursaphelenchus mucronatus genome (Leroy et al., 2003). The variation in genome size across the phylum is probably even larger, since sizes have only been estimated for approximately 50 species (Leroy et al., 2003; Gregory, 2005, Animal Genome Size Databse).
Nematode genomes are similar in size to those of flatworms, annelids, and insects (~60-100 Mb upwards), but are smaller than those of some invertebrates such as molluscs and echinoderms (~400-500 Mb upwards; Gregory, 2005, Animal Genome Size Database). The compact nature of nematode genomes may due to a high rate of large, spontaneous deletions (Witherspoon and Robertson, 2003), and perhaps to selection for deletions (Denver et al., 2004).
The C. briggsae genome is slightly (~4 Mb) larger than the C. elegans genome, due to a larger amount of repetitive DNA in the C. briggsae genome (Stein et al., 2003). This must be due to proliferation of repeat families in the C. briggsae genome, or loss of repetitive DNA from C. elegans. Comparison of the C. elegans and C. briggsae genomes to those of closely related nematodes will shed light on the relative importance of deletions (which will decrease the genome size), versus insertions and proliferation of repeats (which will both increase the genome size).
|
Figure 1. Genome size and chromosome number variation across the phylum Nematoda. Species for which whole-genome sequences will soon be available are highlighted in purple. The phylogeny was adapted from De Ley and Blaxter (2002). Estimates of genome size and chromosome number where taken from: Haemonchus contortus:John Gilleard (pers. comm.) and Leroy et al. (2003); Caenorhabditis elegans: Stein et al. (2003); Pristionchus pacificus: Sommer et al. (1996); clade V nematodes: Blaxter (2000); Meloidogyne hapla: David Bird (pers. comm.); Brugia malayi: Ghedin et al. (2004); Trichinella spiralis: Hammond and Bianco (1992) and Mutafova et al. (1982); other species: Leroy et al. (2003).
Lynch and Conery (2003) suggested that species with smaller effective population sizes (a smaller number of individuals that contribute different alleles to the next generation) have larger genomes, because they tend to accumulate repetitive DNA and genomic duplications. Thus, we expect the genome of C. remanei which is currently being sequenced, to be smaller those of C. elegans and C. briggsae, because its effective population size seems to be larger. That is, in a study of two nuclear genes, the diversity in C. elegans and C. briggsae was just 6-13% of the diversity seen in C. remanei (Graustein et al., 2002). The effective population sizes of parasitic nematodes probably depend on those of their hosts, so parasites of herbivores may have larger effective population sizes than parasites of carnivores or omnivores (Lynch and Conery, 2003). Thus, one could speculate that this explains why the sheep parasite Haemonchus contortus has such a small genome (53 Mb) compared to the human parasite Brugia malayi (85–95 Mb) or the pig parasite Trichinella spiralis (240 Mb).
Since the size difference between the 104 Mb C. briggsae and 100 Mb C. elegans genomes is due to repetitive DNA, they both have ~19,500 genes (Stein et al., 2003). The Brugia malayi genome has a similar size to the Caenorhabditis genomes, ~85-95 Mb, and a similar number of genes, ~18,500 genes (Ghedin et al., 2004; Whitton et al., 2004). The Haemonchus contortus genome is just 53 Mb, but it is not yet clear whether it contains half as many genes as C. elegans, or rather has the same number of genes but half as much non-coding DNA.
Most nematodes have haploid chromosome numbers of n=4–12 (Walton, 1959). The karyotypes of just ~300 species have been studied, but nematodes display a lot of karyotypic variation (Špakulová and Casanova, 2004). The lowest haploid number is n=1 in Parascaris univalens, but very high counts are seen in polyploid species in the Tylenchomorpha. For example, the race of Meloidogyne hapla being sequenced is diploid and has n=14, but another race of M. hapla is polyploid with 2n=45–48 (David Bird, pers. comm.; Triantaphyllou, 1984). Many tylenchomorphs including M. hapla, are parthenogens, in which unfertilized eggs develop into new individuals. Animal species that reproduce in this way seem to be susceptible to polyploidization (Otto and Whitton, 2000). The M. hapla race being sequenced has twice as many chromosomes as most rhabditines, so could reveal traces of an ancient genome duplication in the Tylenchomorpha.
In contrast to the tylenchomorphs, most rhabditines have n=5-6 (Blaxter, 2000). Indeed, C. elegans and C. briggsae both have n=6, even though their chromosomes have undergone ~4000 rearrangements since they diverged (Stein et al., 2003). The lack of fissions or fusions suggests that there could be selection for a stable chromosome number in the Rhabditina.
When Stein et al. (2003) compared the genome of C. elegans to that of C. briggsae, they identified ~4800 conserved segments, with an average size of 37 kb. They estimated that there have been 3.6 interchromosomal rearrangements per Mb in the C. briggsae genome (Stein et al., 2003). Thus, an average C. briggsae chromosome of ~10-20 Mb consists of a mosaic of ~35-70 chunks that match several C. elegans chromosomes. However, some of these segments are very small, so it may be possible to detect ancient Caenorhabditis linkage groups by considering just the largest conserved segments. A genetic map for C. briggsae is currently underway, and should allow us to match each C. briggsae chromosome to the C. elegans chromosome(s) with which it shares common ancestry (Robert Waterston, Raymond Miller, Scott Baird and Asif Chinwalla, unpublished data).
Sequencing of random regions of the Pristionchus pacificus and Brugia malayi genomes suggests that despite the frequent occurrence of reciprocal translocations, ancient secernentean linkage groups may still be detectable. In an 11-gene region sequenced from P. pacificus chromosome III, 10/11 genes had orthologs on C. elegans chromosome III (Lee et al., 2003). This led Lee et al. to suggest that P. pacificus chromosome III and C. elegans chromosome III shared a common ancestor. If this is true, there must have been a lot of intrachromosomal rearrangement since just three pairs of the P. pacificus genes are closely linked in C. elegans, but these pairs are scattered over 12 Mb. Whitton et al. (2004) found evidence suggesting that B. malayi chromosmes can be matched to their C. elegans homologs. They sequenced BAC ends containing 8 Mb of Brugia malayi sequence, and found that 60% of the BACs matched the same C. elegans chromosome at both ends. However, large rearrangements seem to have occurred within chromosomes, because the average distance between two matches was 4 Mb.
Each of Caenorhabditis elegans' chromosomes is divided into a repeat-poor "central cluster" that rarely undergoes meiotic exchange, and two repeat-rich "arms" that have a ~7-fold higher recombination rate (Barnes et al., 1995; C. elegans Sequencing Consortium, 1998). Intriguingly, the arms are evolving far more rapidly than the centers of chromosomes, in terms of both substitutions and chromosomal rearrangements such as translocations, inversions, and duplications (C. elegans Sequencing Consortium, 1998; Stein et al., 2003). This may reflect a lower tolerance to mutation in the central clusters, which contain most of the essential genes and operons (Blumenthal et al., 2002; Kamath et al., 2003). Alternatively, the arms may simply have a higher mutation rate, since the high recombination rate may provoke substitutions (Cutter and Payseur, 2003), while the abundance of repeats probably triggers chromosomal rearrangements (Coghlan and Wolfe, 2002).
Analysis of the Pristionchus pacificus genetic map indicates that at least four of the six chromosomes have genetically-defined central clusters and arms (Ralf Sommer, pers. comm.). Are all nematode chromosomes split into genetically defined central clusters and arms, and do the arms always evolve faster than the centers? Comparison of C. elegans chromosomes to those other nematodes will allow us to address this question; as well as to test the interesting hypothesis of Barnes et al. (1995) that an ancestor of C. elegans had smaller chromosomes consisting of just "clusters", which later sprouted arms due to proliferation of repetitive DNA.
Barnes et al. (1995) noticed that the recombination rate in most C. elegans autosomes differs by a factor of ~7–12 between the arms and central clusters. However, in chromosome V, the recombination rate differs by a factor of just four between the arms and cluster. The relatively higher recombination rate in the central cluster of chromosome V may be a cause (or possibly a result) of its "arm-like" characteristics: its high density of gene families (C. elegans Sequencing Consortium, 1998), low number of essential genes (Kamath et al., 2003), scarcity of operons (Blumenthal et al., 2002), abundant species-specific genes (Parkinson et al., 2004), and low probability of sequence matches to Brugia malayi BAC end sequences (Whitton et al., 2004).
There are ~1000 operons in the C. elegans genome, of which 96% are conserved in C. briggsae, far more than expected if selection selection did not act to preserve operons (60%; Stein et al., 2003). Gene order in ~15% of the genome is stabilized by selection against rearrangements of operons, since 15% of C. elegans genes are part of operons (Blumenthal et al., 2002). In fact, operons are concentrated in the central clusters of C. elegans chromosomes, so probably contribute to the lower rearrangement rate in the centers compared to the arms (Blumenthal et al., 2002). One C. elegans operon is conserved in the closely related rhabditine Oscheius (Evans et al., 1997), but at least one C. elegans operon has been broken in Pristionchus pacificus (Lee and Sommer, 2003). Operons probably exist in the Rhabditina, Tylenchina, and Spirurina, since trans-splicing has been observed in Haemonchus contortus, Panagrellus redivivus, Ascaris suum, Anisakis spp. and Brugia malayi (Bektesh et al., 1988; Takacs et al., 1988). Two unresolved questions are whether Trichinella spiralis has trans-splicing and operons, and whether nematode operons are related to those in flatworms (Davis and Hodgson, 1997).
C. elegans chromosomes also contain small clusters of ~2-5 genes that are co-expressed in muscle, even though they do not belong to operons; as well as clusters co-expressed in the germline, intestine, and neurons (Roy et al., 2002, Table 1: intestine=Mountain 08 and neurons=Mountain 06). Furthermore, C. elegans genes seem to be partitioned between chromosomes according to their general role: essential genes are mostly found in the centers of chromosomes I, II and III; while genes involved in worm behavior and morphology are concentrated on chromosomes II and X (Kamath et al., 2003). This organization may permit co-regulation of genes that sit inside the same chromatin domain (Kamath et al., 2003). There may have been selection against rearrangement of one large cluster of C. elegans genes on chromosome II that are expressed during spermatogenesis, since it is part of the largest conserved segment between the C. elegans and C. briggsae (Miller et al., 2004). In contrast, two other spermatogenesis clusters on C. elegans chromosome IV are not found in C. briggsae (Miller et al., 2004). An intriguing possibility is that these two clusters were assembled just in C. elegans, by rearrangements that were adaptive because they permitted co-regulation of reproductive genes (Miller et al., 2004).
HOX genes are transcription factors that are closely clustered in the genomes of most animals (Bürglin, 1994). They control the expression of anterior-posterior patterning along the body axis during early embryogenesis collinearly with their arrangement on the chromosome. The HOX cluster has been conserved in most animal phyla over hundreds of millions of years of evolution (Ferrier and Holland, 2001), but the nematode HOX cluster is surprisingly poorly preserved. The ancestral bilaterian probably had a cluster of nine HOX genes (nine ortholog groups), but all nematodes have lost at least three ortholog groups (Aboobaker and Blaxter, 2003). A further two ortholog groups were lost in the lineage leading to C. elegans, after the Spirurina-Rhabditina-Tylenchina clade diverged from other nematodes. Aboobaker and Blaxter (2003) point out that these two HOX ortholog groups were lost around the time when C. elegans' ancestor switched from a regulative mode of development to a deterministic lineage-driven mode. They suggest that perhaps the transition freed the two HOX ortholog groups from their role in anterior-posterior patterning, making their loss tolerable. Interestingly, the HOX cluster has been broken up in C. elegans: its six HOX genes (belonging to four ortholog groups) are arranged in three pairs scattered over 5 Mb of chromosome III. Trichinella spiralis probably has a regulative mode of development, but it is not yet known whether its HOX genes are clustered. However, even though Brugia malayi has lineage-driven development, most of its HOX cluster seems to have been preserved intact (Aboobaker and Blaxter, 2003).
In Caenorhabditis elegans, sex determination acts through an X-chromosome dosage mechanism: animals with two X chromosomes develop as hermaphrodites, whereas XO animals develop as males (Nicoll et al., 1997). XX/XO sex determination is very common across the Nematoda (Walton, 1940), suggesting that the first nematode possibly had XX/XO sex determination. Even if the C. elegans and Trichinella spiralis XX/XO systems did share common ancestry, the traces will be hard to find, since sex determination pathways and genes are evolving very quickly both in terms of sequence change and gene regulation (reviewed in Haag and Doty, 2005). However, at least one key gene is conserved in the XX/XO sex determination pathways of C. elegans and Pristionchus pacificus (Pires-daSilva and Sommer, 2004), so it should be possible to determine whether the P. pacificus, C. elegans and Haemonchus contortus XX/XO systems are orthologous.
Only a handful of nematodes have Y chromosomes: Brugia malayi (Underwood and Bianco, 1999), Onchocerca volvulus (Hirai et al., 1987), Baylisascaris transfuga (Mutafova, 1995), Contracaecum incurvum (White, 1973), and Trichuris muris (Špakulová et al., 1994). Since Ys are only known in these few distantly related nematodes, White (1973) suggested that they probably emerged recently. In papaya the sequence of the Y chromosome betrays its recent origin from autosomes (Liu et al., 2004), and it will be interesting to see if the Brugia malayi Y arose in a similar way.
The involvement of the C. elegans X chromosome in sex determination may have restrained its pace of structural evolution. Since C. elegans diverged from C. briggsae, its X chromosome has undergone about half as many rearrangements as its autosomes (Stein et al., 2003). Indeed, two of the three largest conserved segments between the two genomes are on C. elegans X (Stein et al., 2003). Furthermore, a genetic linkage map of Pristionchus pacificus suggests that the X chromosome may have been preserved largely intact since the divergence of P. pacificus from C. elegans (Lee et al., 2003; Ralf Sommer, pers. comm.). Thus, although the genes involved in sex determination tolerate high substitution rates, structural rearrangements of the sex chromosome may be more detrimental to the mechanism of sex determination. A possible reason for a lower rate of rearrangement of the X chromosome compared to autosomes is selection against translocations of the regions of the X chromosome from which dosage compensation is initiated (Csankovszki et al., 2004), and/or of regions that contain the "X-signal elements" whose dosage determines the sex of an embryo (Meyer, 2000). X-autosomal translocations may also be deleterious because they upset the dosage of the translocated genes (Ohno, 1967).
In nematode species in which the Y chromosome determines sex, the rate of chromosomal rearrangement in X may be similar to that in autosomes, or perhaps even higher. In Brugia malayi, which has a Y chromosome, the X chromosome seems to have undergone more interchromosomal rearrangements than the autosomes. That is, among B. malayi BACs that match the C. elegans X chromosome at one end, an unusually high number match an autosome at the other end (Whitton et al., 2004). Perhaps the emergence of a Y chromosome in B. malayi has liberated its X from any function in sex determination, resulting in relaxation of selection against rearrangements of X.
The most common reproductive strategy among nematodes is sexual reproduction between males and females (amphimixis), which is seen in Caenorhabditis remanei, Caenorhabditis sp. PB2801, Caenorhabditis japonica, Haemonchus contortus, Brugia malayi, and Trichinella spiralis. However, alternative reproductive strategies have arisen in some nematode groups, including hermaphroditism, parthenogenesis, and haplo-diploidy. For example, C. elegans, C. briggsae and Pristionchus pacificus are hermaphroditic (see The evolution of nematode sex determination), while the strain of Meloidogyne hapla being sequenced is a facultative meiotic parthenogen (David Bird, pers. comm.). Because hermaphroditic species (and perhaps parthenogenetic species: Archetti, 2004) have a smaller effective population size than amphimictic species, they will tend to accumulate deleterious mutations, resulting in a faster substitution rate and rate of chromosomal rearrangement (Charlesworth, 1992; Cutter and Payseur, 2003). This may explain why substitution rates in C. elegans and Meloidogyne seem to be high compared to most nematodes (Blaxter et al., 1998). We will soon be able to test whether they also have an accelerated rate of chromosomal rearrangement.
Since C. elegans and C. briggsae diverged, their chromosomes have been splintered by ~250 reciprocal translocations, ~1400 inversions and ~2700 transpositions (Stein et al., 2003). Intrachromosomal rearrangement is about four times more frequent than interchromosomal rearrangement. Even so, translocations are surprisingly common in Caenorhabditis compared to flies, in which translocations are extremely rare (Ranz et al., 2001; Sharakhov et al., 2002). This may be because almost all dipterans have "monocentric" chromosomes, in which the kinetochores assemble on a localized region in each chromosome. In contrast, "holocentric" species such as C. elegans and C. briggsae have diffuse kinetochores that form along the length of their chromosomes during mitosis. Since the kinetochores are the primary chromosomal attachment site for spindle microtubules, they play a key role in ensuring high fidelity chromosome transmission in both monocentric and holocentric species. However, little is known of the relationship between the distribution of kinetic activity along chromosomes and the pattern of chromosomal rearrangement. In species with monocentric chromosomes, many translocations will be lethal because they will give rise to acentric or dicentric chromosomes; while species with holocentric chromosomes may be more tolerant of translocations (Dernburg, 2001). Most nematodes have holocentric chromosomes, but Trichinella spiralis and some other trichinellids have monocentric chromosomes (Mutafova et al., 1982; Špakulová et al., 1994). Thus, comparison of the T. spiralis genome to that of C. elegans may provide clues as to whether holocentric chromosomes are more susceptible to rearrangement, and whether the first nematode had holocentric chromosomes.
Parkinson et al. (2004) sequenced ESTs from 30 different nematode species across the phylum, and defined ~94,000 genes from ~60,000 families. Surprisingly, only about 15,000 (15%) of the ~94,000 genes are found in all four clades of nematodes studied (Rhabditina, Tylenchina, Spirurina, Dorylaimia). These 15,000 genes are probably involved in core metabolic or structural pathways, since most of them (91%) have sequence matches outside the Nematoda. In addition, they identified ~1300 genes that are nematode-specific but that are found in most nematodes. These ~1300 genes probably have roles that are important for nematode body plan and life history, and so may shed light on the early evolution of the phylum.
Since C. elegans has diverged from C. briggsae, chemoreceptors have proliferated in the C. elegans genome, so that it now has almost twice as many as C. briggsae (718 versus 429; Stein et al., 2003). Duplication and divergence of extra chemoreceptors may have allowed C. elegans to adopt a slightly different ecological niche than C. briggsae, since it uses chemoreceptors to find food, and to avoid predators, pathogens and toxins (Troemel, 1999). On the other hand, C. elegans seems to have lost several genes (~30 genes) that are found in both Pristionchus pacificus and Haemonchus contortus (Parkinson et al., 2004). For example, C. elegans has lost a DNA methyltransferase gene that is found in P. pacificus — a loss that probably led to the abolition of DNA methylation in C. elegans (Gutierrez and Sommer, 2004). Contrasting the gene families that have been duplicated or lost in each of the ten nematode genomes may reveal selection for different gene contents in different species.
C. elegans has ~1000 genes that are not found in C. briggsae, and that lack any match in sequence databases (Stein et al., 2003). Of these, ~200 have been confirmed by EST or cDNA data, so are definitely not gene prediction errors. These genes may have diverged so rapidly that their C. briggsae homolog is unrecognizable; or may have been assembled de novo via chromosomal rearrangements in the C. elegans genome (Long, 2001). Duplications, chromosomal rearrangements and transposable elements are known to play a role in the birth of novel genes (Betran and Long, 2002; Ganko et al., 2003; Long, 2001). Thus, the abundance of species-specific genes in the arms of C. elegans chromosomes probably results from the arms' high rate of rearrangement (Stein et al., 2003).
C. elegans is not alone in having so many species-specific genes. In a survey of ESTs from 30 nematode species, as many as 30-50% of genes in each species seemed to be species-specific (Parkinson et al., 2004). Among the nematodes whose genomes are being sequenced, the fraction ranged from 19% for Haemonchus contortus to 37% for Trichinella spiralis. This is far higher than the fraction of C. elegans genes that are species-specific (5%). However, some of the putative species-specific T. spiralis genes may not be truly species-specific, but rather are just very divergent; if so, we might be able to identify their C. elegans orthologs by using synteny information. Furthermore, some T. spiralis EST consensus sequences were probably too short to find sequences matches; we may be able to detect C. elegans orthologs of these T. spiralis genes once we know their full-length sequences.
Nematode genespace is probably far larger than 60,000 gene families. Firstly, Parkinson et al.'s survey probably missed many genes expressed at low levels and genes whose expression is restricted to a small number of specialized cells, which are difficult to detect in an EST survey. Secondly, they only sampled 30 species out of at least 25,000 species. Furthermore, all of the species sampled so far are terrestrial, and do not yet include any of the large number of poorly sampled enoplid and chromadorid marine nematodes (Lambshead, 1993). Since the first nematode was probably a marine animal (Poinar, 1983), perhaps an enoplid (Blaxter, 2003), even more diversity in gene content may be found in the oceans.
Horizontal gene transfer occurs frequently in prokaryotes, but seems to be rare in eukaryotes. For example, ~1% of the gene repertoire in the nematode Meloidogyne probably originated by horizontal transfer (Scholl et al., 2003), compared to 1-5% of single-copy genes and at least 22% of gene duplicates in Y-Proteobacteria (Lerat et al., 2005) Meloidogyne hapla, a plant parasitic nematode, seems to have gained at least a dozen genes by horizontal gene transfer from bacteria that occupy similar niches in the soil and roots (Scholl et al., 2003). Those genes gained are useful for the nematode's parasitic lifestyle, such as cellulases for digesting plant material, and signaling molecules that induce morphological changes in the plant, facilitating invasion (McCarter et al., 2003; Weerasinghe et al., 2005). A distantly related plant parasite, Bursaphelenchus xylophilus, seems to have independently acquired a cellulase gene from a fungus (Kikuchi et al., 2004). Perhaps horizontal transfer can spur the transition to parasitism (Weerasinghe et al., 2005)?
Several groups of parasitic nematodes, including Brugia malayi, live in symbiosis with specific bacteria carried by the nematodes (see Table 1 in Blaxter, 2003). Some of these are extracellular symbionts, but others are intracellular, such as Wolbachia living in B. malayi and other filarial nematodes. The capture of the Wolbachia gene set seems to have been adaptive for filarial nematodes, since killing Wolbachia with antibiotics reduces the growth and fecundity of the nematodes (Foster et al., 2005; Hoerauf et al., 2001).
Free-living nematodes may also have pinched genes from organisms that live nearby Many nematodes use other animals, often arthropods and molluscs, as transport hosts. For example, C. remanei lives in close association with molluscs and isopods (Baird, 1999). Indeed, C. elegans has four genes, including an alcohol dehydrogenase, that have stronger sequence matches to fungi than to other animals (Parkinson and Blaxter, 2003). These C. elegans genes group with fungal genes in phylogenetic trees. Similar phylogenetic analyses will allow us to scan the eight new genomes for stolen genes.
Parasitism of plants and animals has evolved independently at least nine times in the history of the nematodes (Dorris et al., 1999). Four of the nematodes whose genomes are being sequenced are parasites: Haemonchus contortus, Meloidogyne hapla, Brugia malayi and Trichinella spiralis. The adoption of parasitism in nematodes probably required adaptation of genes present in their free-living ancestors (Blaxter, 2003). For example, modification of nutrient-acquisition genes found in C. elegans, such as digestive enzymes or secreted hydrolases, are likely to have been important for the evolution of parasitism (Geary and Thompson, 2001). The ability of parasitic nematodes to survive immunological attack, some living in an infected individual for years, has long been a puzzle. The cuticle is the main site of interaction between a nematode and its environment, and many nematode genes so far implicated in evading host defenses are secreted or cuticle proteins (Davis et al., 2004; Maizels et al., 2001). For example, the main soluble surface glycoprotein of filarial nematodes, a secreted glutathione peroxidase (GPX-1), is hypothesized to have a role in immune evasion (Zvelebil et al., 1993). In viral, bacterial and protozoan parasites, genes involved in host immune evasion or recognition are often under positive selection, and so show patterns of rapid amino acid substitution (McInerney et al., 2003). Indeed, B. malayi GPX-1 shows signs of positive selection (Zvelebil et al., 1993). By scanning for Haemonchus contortus genes that have diverged sharply in sequence from their Pristionchus and Caenorhabditis orthologs, and that bear secretory signals (Harcus et al., 2004), it may be possible to identify H. contortus genes that have adapted for a parasitic lifestyle.
Some genes essential for parasitism in worms may be novel genes. One possible source is gene duplication, which allows one duplicate to keep the original role, and the other duplicate to take on a parasitic role. For example, the alt gene family of filarial nematodes, which has been implicated in establishing infection, has a single C. elegans ortholog (Gomez-Escobar et al., 2002). On the other hand, other novel genes adapted for parasitism may have been assembled de novo, or have been gained by horizontal gene transfer: plant parasitic nematodes seem to have acquired "parasitism genes" from bacteria in their environment (Bird et al., 2003).
Some "parasitism genes" may by identifiable by examining the expression pattern of their C. elegans orthologs. In Haemonchus contortus and Brugia malayi, the infective stage of the life cycle is the third larval (L3) developmental stage (Blaxter, 2003). In C. elegans the L3 developmental stage is an alternative developmental pathway adopted when food is scarce, called the dauer larva. Thus, identifying the orthologs of C. elegans genes expressed in the dauer larva (Wang and Kim, 2003) may be a route to pinpointing Brugia and Haemonchus genes involved in infection (Bürglin et al., 1998).
Mushegian et al. (1998) compared 36 C. elegans and Drosophila protein orthologs to their yeast counterparts, and found that many C. elegans genes have evolved twice as fast as their Drosophila orthologs. Nematode rRNA genes also seem to have a substitution rate that is 2-3 times that of other animal phyla (Aguinaldo et al., 1997). For example, the rRNA gene divergence between Caenorhabditis species is comparable to that between vertebrate species (The phylogenetic relationships of C. elegans and other Rhabditids; Kiontke et al., 2004)!
To accurately estimate the evolutionary rate in nematodes, ideally we would divide the number of mutations between two closely related species by their divergence date. However, estimating divergence dates between nematode species is extremely difficult, because nematode fossils are scarce (Poinar, 1983). One solution is to calibrate the molecular clock using the date that nematodes diverged from other animals. This allows evolutionary rates and speciation dates to be estimated from the subset of genes that have evolved at the same rate in all animals (for example, see Stein et al., 2003). Unfortunately, estimates made using this approach have large margins of error, due to uncertainties in the date of divergence of nematodes from other animals (estimates range from 600-1300 million years ago: Benton and Ayala, 2003; Blair and Hedges, 2005; Hedges et al., 2004; Peterson et al., 2004), and in the relationship of nematodes to other animals (Philip et al., 2005; Philippe et al., 2005). Ongoing whole-genome sequencing projects for members of eight more animal phyla (Bernal et al., 2001: http://www.genomesonline.org/) may lead to a more accurate phylogenetic tree of animal phyla, reducing these sources of error.
Another solution is to directly measure the mutation rate in laboratory animals. In an ingenious experiment, Denver et al. (2004) estimated the mutation rate in C. elegans by sequencing random stretches of DNA in mutation accumulation lines. They estimated the genomic mutation rate to be ~2.1 mutations per genome per generation. Of the mutations observed, 43% were substitutions, 43% insertions, and 13% deletions. Comparable estimates for other animals do not yet exist, as estimates using earlier techniques were probably less accurate (reviewed by Keightley and Charlesworth, 2005). The future use of this method for detecting mutations in arthropods and vertebrates will provide insight into the difference between the rate and types of mutations in nematodes and that in other animals.
Many mysteries remain in eukaryotic genome evolution. We will soon have a data set of ten nematode genome sequences that will be ideal for investigating unresolved questions, such as what are the forces governing the evolution of chromosome number, size and structure; how does sex chromosome evolution differ from that of autosomes; how do differences in life history traits and reproductive strategy affect genome evolution; and what are the major genomic changes that enable species to adapt to new ecological niches such as parasitism. Looking forward, it seems very possible that once again these tiny animals will be first in revealing some of nature’s deepest secrets.
I am very grateful to David Bird, John Speith, Ralf Sommer, John Gilleard, Robert Waterston, Raymond Miller and David Baillie for useful information and discussion. I am grateful to David Fitch and two anonymous reviewers for their very helpful comments on the text. I thank Richard Durbin and Des Higgins for generously allowing me to complete this work in their labs. Avril Coghlan is supported by the Wellcome Trust.
Aboobaker, A., and Blaxter, M. (2003a). Hox gene evolution in nematodes: novelty conserved. Curr. Opin. Genet. Dev. 13, 593–598. Abstract Article
Aboobaker, A.A., and Blaxter, M.L. (2003b). Hox gene loss during dynamic evolution of the nematode cluster. Curr. Biol. 13, 37–40. Article
Aguinaldo, A.M., Turbeville, J.M., Linford, L.S., Rivera, M.C., Garey, J.R., Raff, R.A., and Lake, J.A. (1997). Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387, 489–493. Abstract Article
Andrássy, I., and Zombori, L. (1976). Evolution as a basis for the systematization of nematodes. (London: Pitman).
Archetti, M. (2004). Recombination and loss of complementation: a more than two-fold cost for parthenogenesis. J. Evol. Biol. 17, 1084–1097. Abstract Article
Baird, S.E. (1999). Natural and experimental association of Caenorhabditis remanei with Trachelipus rathkii and other terrestrial isopods. Nematology 1, 471–475.
Barnes, T.M., Kohara, Y., Coulson, A., and Hekimi, S. (1995). Meiotic recombination, noncoding DNA and genomic organization in Caenorhabditis elegans Genetics 141, 159–179. Abstract Article
Bektesh, S., Van Doren, K., and Hirsh, D. (1988). Presence of the Caenorhabditis elegans spliced leader on different mRNAs and in different genera of nematodes. Genes Dev. 2, 1277–1283. Abstract
Benton, M.J., and Ayala, F.J. (2003). Dating the tree of life. Science 300, 1698–1700. Abstract Article
Bernal, A., Ear, U., and Kyrpides, N. (2001). Genomes OnLine Database (GOLD): a monitor of genome projects world-wide. Nucleic Acids Res. 29, 126–127. Abstract Article
Betran, E., and Long, M. (2002). Expansion of genome coding regions by acquisition of new genes. Genetica 115, 65–80. Abstract Article
Bird, D.M., Opperman, C.H., and Davies, K.G. (2003). Interactions between bacteria and plant-parasitic nematodes: now and then. Int. J. Parasitol. 33, 1269–1276. Abstract Article
Blair, J.E., and Hedges, S.B. (2005). Molecular clocks do not support the Cambrian explosion. Mol. Biol. Evol. 22, 387–390. Abstract Article
Blaxter, M. (2000). Genes and genomes of Necator americanus and related hookworms. Int. J. Parasitol. 30, 347–355. Abstract Article
Blaxter, M.L. (2003). Nematoda: genes, genomes and the evolution of parasitism. Adv. Parasitol. 54, 101–195. Abstract Article
Blaxter, M.L., De Ley, P., Garey, J.R., Liu, L.X., Scheldeman, P., Vierstraete, A., Vanfleteren, J.R., Mackey, L.Y., Dorris, M., Frisse, L.M., et al. (1998). A molecular evolutionary framework for the phylum Nematoda. Nature 392, 71–75. Abstract Article
Blumenthal, T., Evans, D., Link, C.D., Guffanti, A., Lawson, D., Thierry-Mieg, J., Thierry-Mieg, D., Chiu, W.L., Duke, K., Kiraly, M., et al. (2002). A global analysis of Caenorhabditis elegans operons. Nature 417, 851–854. Abstract Article
Bürglin, T.R. (1994). A comprehensive classification of homeobox genes. In Guidebook to the homeobox genes, D. Duboule, ed. (Oxford, United Kingdom: Oxford University Press), pp. 25–71.
Bürglin, T.R., Lobos, E., and Blaxter, M.L. (1998). Caenorhabditis elegans as a model for parasitic nematodes. Int. J. Parasitol. 28, 395–411. Abstract Article
C. elegans Sequencing Consortium (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018. Abstract Article
Charlesworth, B. (1992). Evolutionary rates in partially self-fertilizing species. Am. Nat. 140, 126–148. Article
Coghlan, A., and Wolfe, K.H. (2002). Fourfold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res. 12, 857–867. Abstract Article
Csankovszki, G., McDonel, P., and Meyer, B.J. (2004). Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science 303, 1182–1185. Abstract Article
Cutter, A.D., and Payseur, B.A. (2003a). Rates of deleterious mutation and the evolution of sex in Caenorhabditis. J. Evol. Biol. 16, 812–822. Abstract Article
Cutter, A.D., and Payseur, B.A. (2003b). Selection at linked sites in the partial selfer Caenorhabditis elegans. Mol. Biol. Evol. 20, 665–673. Abstract Article
Davis, E.L., Hussey, R.S., and Baum, T.J. (2004). Getting to the roots of parasitism by nematodes. Trends Parasitol. 20, 134–141. Abstract Article
Davis, R.E., and Hodgson, S. (1997). Gene linkage and steady state RNAs suggest trans-splicing may be associated with a polycistronic transcript in Schistosoma mansoni. Mol. Biochem. Parasitol. 89, 25–39. Abstract Article
De Ley, P., and Blaxter, M.L. (2002). Systematic position and phylogeny. In The Biology of Nematodes, D. Lee, ed. (Reading: Harwood Academic Publishers).
Denver, D.R., Morris, K., Lynch, M., and Thomas, W.K. (2004). High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430, 679–682. Abstract Article
Dernburg, A.F. (2001). Here, there, and everywhere: kinetochore function on holocentric chromosomes. J. Cell. Biol. 153, F33–F38. Abstract Article
Dorris, M., De Ley, P., and Blaxter, M.L. (1999). Molecular analysis of nematode diversity and the evolution of parasitism. Parasitol Today 15, 188–193. Abstract Article
Evans, D., Zorio, D., MacMorris, M., Winter, C.E., Lea, K., and Blumenthal, T. (1997). Operons and SL2 trans-splicing exist in nematodes outside the genus Caenorhabditis. Proc. Natl. Acad. Sci. USA 94, 9751–9756. Abstract
Ferrier, D.E., and Holland, P.W. (2001). Ancient origin of the Hox gene cluster. Nat. Rev. Genet. 2, 33–38. Abstract Article
Foster, J., Ganatra, M., Kamal, I., Ware, J., Makarova, K., Ivanova, N., Bhattacharyya, A., Kapatral, V., Kumar, S., Posfai, J., et al. (2005). The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS. Biol. 3, e121. Abstract Article
Ganko, E.W., Bhattacharjee, V., Schliekelman, P., and McDonald, J.F. (2003). Evidence for the contribution of LTR retrotransposons to C. elegans gene evolution. Mol. Biol. Evol. 20, 1925–1931. Abstract Article
Geary, T.G., and Thompson, D.P. (2001). Caenorhabditis elegans: how good a model for veterinary parasites? Vet. Parasitol. 101, 371–386. Abstract Article
Ghedin, E., Wang, S., Foster, J.M., and Slatko, B.E. (2004). First sequenced genome of a parasitic nematode. Trends Parasitol. 20, 151–153. Abstract Article
Gomez-Escobar, N., Gregory, W.F., Britton, C., Murray, L., Corton, C., Hall, N., Daub, J., Blaxter, M.L., and Maizels, R.M. (2002). Abundant larval transcript-1 and -2 genes from Brugia malayi: diversity of genomic environments but conservation of 5' promoter sequences functional in Caenorhabditis elegans. Mol. Biochem. Parasitol. 125, 59–71. Abstract Article
Graustein, A., Gaspar, J.M., Walters, J.R., and Palopoli, M.F. (2002). Levels of DNA polymorphism vary with mating system in the nematode genus Caenorhabditis. Genetics 161, 99–107. Abstract
Gutierrez, A., and Sommer, R.J. (2004). Evolution of dnmt-2 and mbd-2-like genes in the free-living nematodes Pristionchus pacificus, Caenorhabditis elegans and Caenorhabditis briggsae. Nucleic Acids Res. 32, 6388–6396. Abstract Article
Haag, E.S., and Doty, A.V. (2005). Sex determination across evolution: connecting the dots. PLoS. Biol. 3, e21. Abstract Article
Hammond, M.P., and Bianco, A.E. (1992). Genes and genomes of parasitic nematodes. Parasitol Today 8, 299–305. Abstract Article
Harcus, Y.M., Parkinson, J., Fernandez, C., Daub, J., Selkirk, M.E., Blaxter, M.L., and Maizels, R.M. (2004). Signal sequence analysis of expressed sequence tags from the nematode Nippostrongylus brasiliensis and the evolution of secreted proteins in parasites. Genome Biol. 5, R39. Abstract Article
Hedges, S.B., Blair, J.E., Venturi, M.L., and Shoe, J.L. (2004). A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol. Biol. 4, 2. Abstract Article
Hirai, H., Tada, I., Takahashi, H., Nwoke, B.E., and Ufomadu, G.O. (1987). Chromosomes of Onchocerca volvulus (Spirurida: Onchocercidae): a comparative study between Nigeria and Guatemala. J. Helminthol. 61, 43–46. Abstract
Hoerauf, A., Mand, S., Adjei, O., Fleischer, B., and Buttner, D.W. (2001). Depletion of Wolbachia endobacteria in Onchocerca volvulus by doxycycline and microfilaridermia after ivermectin treatment. Lancet 357, 1415–1416. Abstract Article
Kamath, R.S., Fraser, A.G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. Abstract Article
Keightley, P.D., and Charlesworth, B. (2005). Genetic instability of C. elegans comes naturally. Trends Genet. 21, 67–70. Abstract Article
Kikuchi, T., Jones, J.T., Aikawa, T., Kosaka, H., and Ogura, N. (2004). A family of glycosyl hydrolase family 45 cellulases from the pine wood nematode Bursaphelenchus xylophilus. FEBS Lett. 572, 201–205. Abstract Article
Kiontke, K., Gavin, N.P., Raynes, Y., Roehrig, C., Piano, F., and Fitch, D.H. (2004). Caenorhabditis phylogeny predicts convergence of hermaphroditism and extensive intron loss. Proc. Natl. Acad. Sci. USA 101, 9003–9008. Abstract Article
Lambshead, P.J. (1993). Recent developments in marine benthic biodiversity research. Oceanis 19, 5–24.
Lee, K.Z., Eizinger, A., Nandakumar, R., Schuster, S.C., and Sommer, R.J. (2003). Limited microsynteny between the genomes of Pristionchus pacificus and Caenorhabditis elegans. Nucleic Acids Res. 31, 2553–2560. Abstract Article
Lee, K.Z., and Sommer, R.J. (2003). Operon structure and trans-splicing in the nematode Pristionchus pacificus. Mol. Biol. Evol. 20, 2097–2103. Abstract Article
Lerat, E., Daubin, V., Ochman, H., and Moran, N.A. (2005). Evolutionary origins of genomic repertoires in bacteria. PLoS. Biol. 3, e130. Abstract Article
Leroy, S., Duperray, C., and Morand, S. (2003). Flow cytometry for parasite nematode genome size measurement. Mol. Biochem. Parasitol. 128, 91–93. Abstract Article
Liu, Z., Moore, P.H., Ma, H., Ackerman, C.M., Ragiba, M., Yu, Q., Pearl, H.M., Kim, M.S., Charlton, J.W., Stiles, J.I., et al. (2004). A primitive Y chromosome in papaya marks incipient sex chromosome evolution. Nature 427, 348–352. Abstract Article
Lynch, M., and Conery, J.S. (2003). The origins of genome complexity. Science 302, 1401–1404. Abstract Article
Maizels, R.M., Blaxter, M.L., and Scott, A.L. (2001). Immunological genomics of Brugia malayi: filarial genes implicated in immune evasion and protective immunity. Parasite Immunol. 23, 327–344. Abstract Article
McInerney, J.O., Littlewood, D.T., and Creevey, C.J. (2003). Detecting adaptive molecular evolution: additional tools for the parasitologist. Adv. Parasitol. 54, 359–379. Abstract Article
Meyer, B.J. (2000). Sex in the wormcounting and compensating X-chromosome dose. Trends Genet. 16, 247–253. Abstract Article
Miller, M.A., Cutter, A.D., Yamamoto, I., Ward, S., and Greenstein, D. (2004). Clustered organization of reproductive genes in the C. elegans genome. Curr. Biol. 14, 1284–1290. Abstract Article
Mushegian, A.R., Garey, J.R., Martin, J., and Liu, L.X. (1998). Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 8, 590–598. Abstract
Mutafova, T. (1995). Meiosis and some aspects of cytological mechanisms of chromosomal sex determination in nematode species. Int. J. Parasitol. 25, 453–462. Abstract Article
Mutafova, T., Dimitrova, Y., and Komandarev, S. (1982). The karyotype of four Trichinella species. Z. Parasitenkd. 67, 115–120. Abstract Article
Nicoll, M., Akerib, C.C., and Meyer, B.J. (1997). X-chromosome-counting mechanisms that determine nematode sex. Nature 388, 200–204. Abstract Article
Niedermaier, J., and Moritz, K.B. (2000). Organization and dynamics of satellite and telomere DNAs in Ascaris: implications for formation and breakdown of compound chromosomes. Chromosoma 109, 439–452. Abstract
Ohno, S. (1967). Sex chromosomes and sex-linked genes. In: Monographs on endocrinology, A. Labhart, ed. (Heidelberg: Springer-Verlag), pp. 123–135.
Otto, S.P., and Whitton, J. (2000). Polyploid incidence and evolution. Annu. Rev. Genet. 34, 401–437. Abstract Article
Parkinson, J., and Blaxter, M. (2003). SimiTri-visualizing similarity relationships for groups of sequences. Bioinformatics 19, 390–395. Abstract Article
Parkinson, J., Mitreva, M., Whitton, C., Thomson, M., Daub, J., Martin, J., Schmid, R., Hall, N., Barrell, B., Waterston, R.H., et al. (2004). A transcriptomic analysis of the phylum Nematoda. Nat. Genet. 36, 1259–1267. Abstract Article
Peterson, K.J., Lyons, J.B., Nowak, K.S., Takacs, C.M., Wargo, M.J., and McPeek, M.A. (2004). Estimating metazoan divergence times with a molecular clock. Proc. Natl. Acad. Sci. USA 101, 6536–6541. Abstract Article
Philip, G.K., Creevey, C.J., and McInerney, J.O. (2005). The Opisthokonta and the Ecdysozoa may not be clades: stronger support for the grouping of plant and animal than for animal and fungi and stronger support for the Coelomata than Ecdysozoa. Mol. Biol. Evol. Abstract Article
Philippe, H., Lartillot, N., and Brinkmann, H. (2005). Multigene analyses of bilaterian animals corroborate the monophyly of Ecdysozoa, Lophotrochozoa and Protostomia. Mol. Biol. Evol. Abstract Article
Pires-daSilva, A., and Sommer, R.J. (2004). Conservation of the global sex determination gene tra-1 in distantly related nematodes. Genes Dev. 18, 1198–1208. Abstract Article
Platt, H.M. (1994). Foreward. In: The phylogenetic systematics of free-living nematodes, S. Lorenzen, ed. (London: The Ray Society).
Ranz, J.M., Casals, F., and Ruiz, A. (2001). How malleable is the eukaryotic genome? Extreme rate of chromosomal rearrangement in the genus Drosophila. Genome Res. 11, 230–239. Abstract Article
Roy, P.J., Stuart, J.M., Lund, J., and Kim, S.K. (2002). Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418, 975–979. Abstract Article
Scholl, E.H., Thorne, J.L., McCarter, J.P., and Bird, D.M. (2003). Horizontally transferred genes in plant-parasitic nematodes: a high-throughput genomic approach. Genome Biol. 4, R39. Abstract Article
Sharakhov, I.V., Serazin, A.C., Grushko, O.G., Dana, A., Lobo, N., Hillenmeyer, M.E., Westerman, R., Romero-Severson, J., Costantini, C., Sagnon, N., et al. (2002). Inversions and gene order shuffling in Anopheles gambiae and A. funestus. Science 298, 182–185. Abstract Article
Sommer, R.J., Carta, L.K., Kim, S.-Y., and Sternberg, P.W. (1996). Morphological, genetic and molecular description of Pristionchus pacificus sp. n. (Nematoda: Neodiplogastridae). Fundam. Appl. Nematol. 19, 511–521.
Špakulová, M., and Casanova, J.C. (2004). Current knowledge on B chromosomes in natural populations of helminth parasites: a review. Cytogenet. Genome Res. 106, 222–229. Abstract Article
Špakulová, M., Král'ová, I., and Cutillas, C. (1994). Studies on the karyotype and gametogenesis in Trichuris muris. J. Helminthol. 68, 67–72. Abstract
Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. (2003). The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS. Biol. 1, E45. Abstract Article
Takacs, A.M., Denker, J.A., Perrine, K.G., Maroney, P.A., and Nilsen, T.W. (1988). A 22-nucleotide spliced leader sequence in the human parasitic nematode Brugia malayi is identical to the trans-spliced leader exon in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 85, 7932–7936. Abstract
Triantaphyllou, A.C. (1984). Polyploidy in meiotic parthenogenetic populations of Meloidogyne hapla and a mechanism of conversion to diploidy. Rev. Nematol. 7, 65–72.
Troemel, E.R. (1999). Chemosensory signaling in C. elegans. Bioessays 21, 1011–1020. Abstract
Underwood, A.P., and Bianco, A.E. (1999). Identification of a molecular marker for the Y chromosome of Brugia malayi. Mol. Biochem. Parasitol. 99, 1–10. Abstract Article
Walton, A.C. (1940). Gametogenesis. In: An Introduction to Nematology, G.B. Chitwood, and M.B. Chitwood, eds. (New York: Babylon), pp. 205–215.
Wang, J., and Kim, S.K. (2003). Global analysis of dauer gene expression in Caenorhabditis elegans. Development 130, 1621–1634. Abstract Article
Weerasinghe, R.R., Bird, D.M., and Allen, N.S. (2005). Root-knot nematodes and bacterial Nod factors elicit common signal transduction events in Lotus japonicus. Proc. Natl. Acad. Sci. USA. Abstract Article
White, M.J.D. (1973). Animal Cytology and Evolution, 3rd edn (New York: Cambridge University Press).
Whitton, C., Daub, J., Quail, M., Hall, N., Foster, J., Ware, J., Ganatra, M., Slatko, B., Barrell, B., and Blaxter, M. (2004). A genome sequence survey of the filarial nematode Brugia malayi: repeats, gene discovery, and comparative genomics. Mol. Biochem. Parasitol. 137, 215–227. Abstract Article
Witherspoon, D.J., and Robertson, H.M. (2003). Neutral evolution of ten types of mariner transposons in the genomes of Caenorhabditis elegans and Caenorhabditis briggsae. J. Mol. Evol. 56, 751–769. Abstract Article
Zvelebil, M.J., Tang, L., Cookson, E., Selkirk, M.E., and Thornton, J.M. (1993). Molecular modelling and epitope prediction of gp29 from lymphatic filariae. Mol. Biochem. Parasitol. 58, 145–153 Abstract
*Edited by David H.A. Fitch. Last revised September 07, 2005. Published September 07, 2005. This chapter should be cited as: Coghlan, A. Nematode genome evolution (September 07, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.15.1, http://www.wormbook.org.
Copyright: © 2005 Avril Coghlan. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: alc@sanger.ac.uk
†Current address: Wellcome Trust Sanger Institute, Cambridge, CB10 1SA, UK
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.