Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
The completion of the C. elegans genome sequence permits the comprehensive examination of the expression and function of genes. Annotation of virtually every encoded gene in the genome allows systematic analysis of those genes using high-throughput assays, such as microarrays and RNAi. This chapter will center on the use of microarrays to comprehensively identify genes with enriched expression in the germ line during development. This knowledge provides a database for further studies that focus on gene function during germline development or early embryogenesis. Additionally, a comprehensive overview of germline gene expression can uncover striking biases in how genes expressed in the germ line are distributed in the genome, leading to new discoveries of global regulatory mechanisms in the germ line.
Regulation of gene expression at both transcriptional and post-transcriptional levels is crucial for the proper specification, proliferation and differentiation of germ cells. At the transcriptional level, certain genes are effectively silenced while other genes are expressed (see Specification of the germ line and Germline chromatin). After transcript production, complex networks of RNA regulatory proteins modulate either the stability and/or translation of mRNAs. These processes combine to effectively control, both spatially and temporally, the levels of germline-expressed gene products. Our understanding of these mechanisms in the germ line is still murky. How are genes selected for silencing or expression? How does germline gene expression influence the organization of genes in the genome, and vice versa? How are the dramatic structural changes of meiotic chromosomes coordinated with continued gene expression? What mechanisms control spatial and temporal expression within the germ line?
For those wishing to understand these aspects of germline development, one goal is to identify every germline-expressed gene. With such information in hand, it is possible to analyze gene functions by RNAi and decipher regulatory mechanisms by a combination of bioinformatics and molecular genetics. Such genome-wide studies reveal global trends, and also identify individual genes that might function to control germ cell fate decisions or terminal differentiation of gametes, making them candidates for future functional study.
DNA microarray technology is frequently used to monitor changes in gene expression for thousands of genes simultaneously (reviewed in Duggan et al., 1999). Typically, DNA microarrays are common laboratory glass slides upon which is printed a series of discrete spots of DNA. Each spot corresponds to some portion of a known gene or predicted open reading frame. For each experiment, a comparison is made between two different populations of transcripts that have been reverse-transcribed to cDNA, each in the presence of a distinct fluorescent label. At each spot, competitive hybridization between the two populations occurs. The relative intensity of the two fluorescent labels at each spot measures the relative abundance of a given gene transcript between the two populations (Figure 1). When used in this manner, microarrays provide information about the steady state levels of mRNAs; they do not directly address the rate at which a transcript is synthesized or degraded. The C. elegans DNA microarrays that primarily have been used to investigate germline gene expression contain over 18,000 spots and represent about 90% of the currently annotated genes in the genome (Jiang et al., 2001; see DNA microarrays).
|
Figure 1. Example of microarray hybridization. A representative portion of a microarray shows the differential signals from two RNA samples. One RNA sample was reverse transcribed into cDNAs labelled with red fluorophore, the other RNA sample into cDNAs labelled with green fluorophore, and the cDNA mixture was hybridized to spots of DNA representing different genes. Selective hybridization of cDNA from either RNA sample to a DNA spot produces red or green signal; hybridization of cDNA from both RNA samples produces yellow signal. In this example, red spots represent RNAs enriched in hermaphrodites with wild type germ lines, and green spots are RNAs enriched in glp-4(bn2) mutants with greatly diminished germ lines.
The comprehensive identification of germline-expressed genes will comprise a molecular definition of germline components, and provide the framework to identify individual genes involved in specific germline functions. Microarrays are an excellent platform to achieve such a goal because the expression of all genes in the genome can be examined simultaneously. Microarray analysis of mutants that perturb specific aspects of germline development has been very useful in defining sets of genes with germline-enriched expression. Discussed below are three sets of genes that have been identified to date: genes with enriched expression in the germline relative to the soma, genes with enriched expression during spermatogenesis, and genes with enriched expression during oogenesis.
One of the most straightforward comparisons identified genes with germline-enriched expression relative to somatic tissues (Reinke et al., 2000; Reinke et al., 2004). A temperature-sensitive mutation, glp-4(bn2), causes severe defects in germ cell proliferation, resulting in gonads with ∼100-fold fewer germ cells than wild type (Beanan and Strome, 1992). Microarray comparison of either L4 or young adult wild type hermaphrodites to glp-4(ts) hermaphrodites of the corresponding stage identified 3144 genes with germline-enriched expression relative to somatic tissues in hermaphrodites (>2x, p<0.01). When a similar comparison was done with wild-type and glp-4(ts) males, 1092 genes showed significantly enriched expression in the germ line. Previous single-gene analyses had identified ∼70 genes that are expressed and/or function in the hermaphrodite germline. Essentially every one of these genes was correctly identified as germline-enriched in the microarray experiments, demonstrating the accuracy and comprehensiveness of the microarray approach. However, it is important to note that a gene with equivalent expression in both samples, even if expressed in the germline, will not be identified as a “germline gene”. Thus, these studies are limited to identification of genes with enriched expression in the germ line relative to the soma.
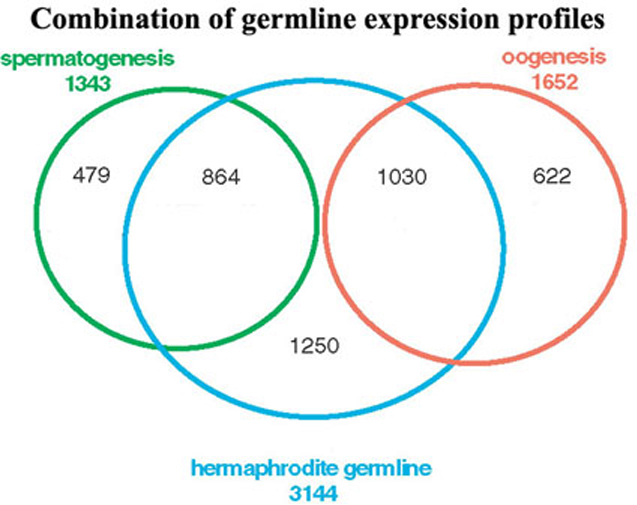
While the identification of genes with germline-enriched expression in the two sexes is informative, the number of genes identified is formidably large. Further microarray experiments using mutants that affect gametogenesis helped to group these genes into distinct categories. fem-1(lf) mutant hermaphrodites are defective in initiation of spermatogenesis and consequently produce only oocytes (Nelson et al., 1978), while fem-3(gf) mutants are defective in switching from spermatogenesis to oogenesis and consequently produce only sperm (Barton et al. 1987). Direct comparison of the genes differentially expressed between fem-1(lf) and fem-3(gf) populations defined two sets of genes: those expressed during spermatogenesis (1343) and those expressed during oogenesis (1652) (>2x, p<0.01). Genes with high expression during spermatogenesis are likely to be involved in spermatocyte specification and differentiation. Genes with high expression during oogenesis are expected to encode proteins required for oocyte differentiation as well as maternally provided factors necessary for proper development of the early embryo.
Combination of the glp-4(ts) vs wild type comparison and the fem-1(lf) vs fem-3(gf) comparison identifies 4245 genes with expression in the germ line and/or gametes – approximately 20% of all the genes in the genome (Figure 2). These genes can be divided into three groups, spermatogenesis (genes in the green circle), oogenesis (genes in the orange circle), and intrinsic, which includes germline genes not differentially expressed during gametogenesis (genes in the blue circle but not in the green or orange circles). Most of the spermatogenesis-enriched genes identified in hermaphrodites were also enriched in males, as expected.
|
Figure 2. Venn diagram of germline gene sets. The wild type vs. glp-4 (diminished germ line) data set was combined with the fem-1(lf) (oocytes only) vs. fem-3(gf) (sperm only) data set to generate the displayed set of genes with germline-enriched expression patterns. Reprinted with permission from Reinke et al., 2004.
In addition to expression profiles of mutants, temporal expression profiles gathered from wild type animals can also reveal sets of co-regulated genes. Microarrays have been probed with RNAs prepared at consecutive times of development from wild type animals. This timecourse provides high temporal resolution (every 3 hours for 36 hours) of gene expression from mid-L3 through adulthood (Reinke et al., 2004). During this period, major germline events include entry into meiosis, spermatogenesis, oogenesis and embryogenesis. Because intact animals were used for the timecourse analysis, fluctuations in expression of somatic genes would also be detected. Among the major developmental events in the soma during this period are gonadogenesis (i.e., generation of uterus, spermathecae and sheath cells) and vulval development. Statistical analysis using ANOVA identified 5083 genes with a significant fluctuation in expression level during the timecourse (p<0.01); among these, 2426 genes had also been identified as germline-regulated genes.
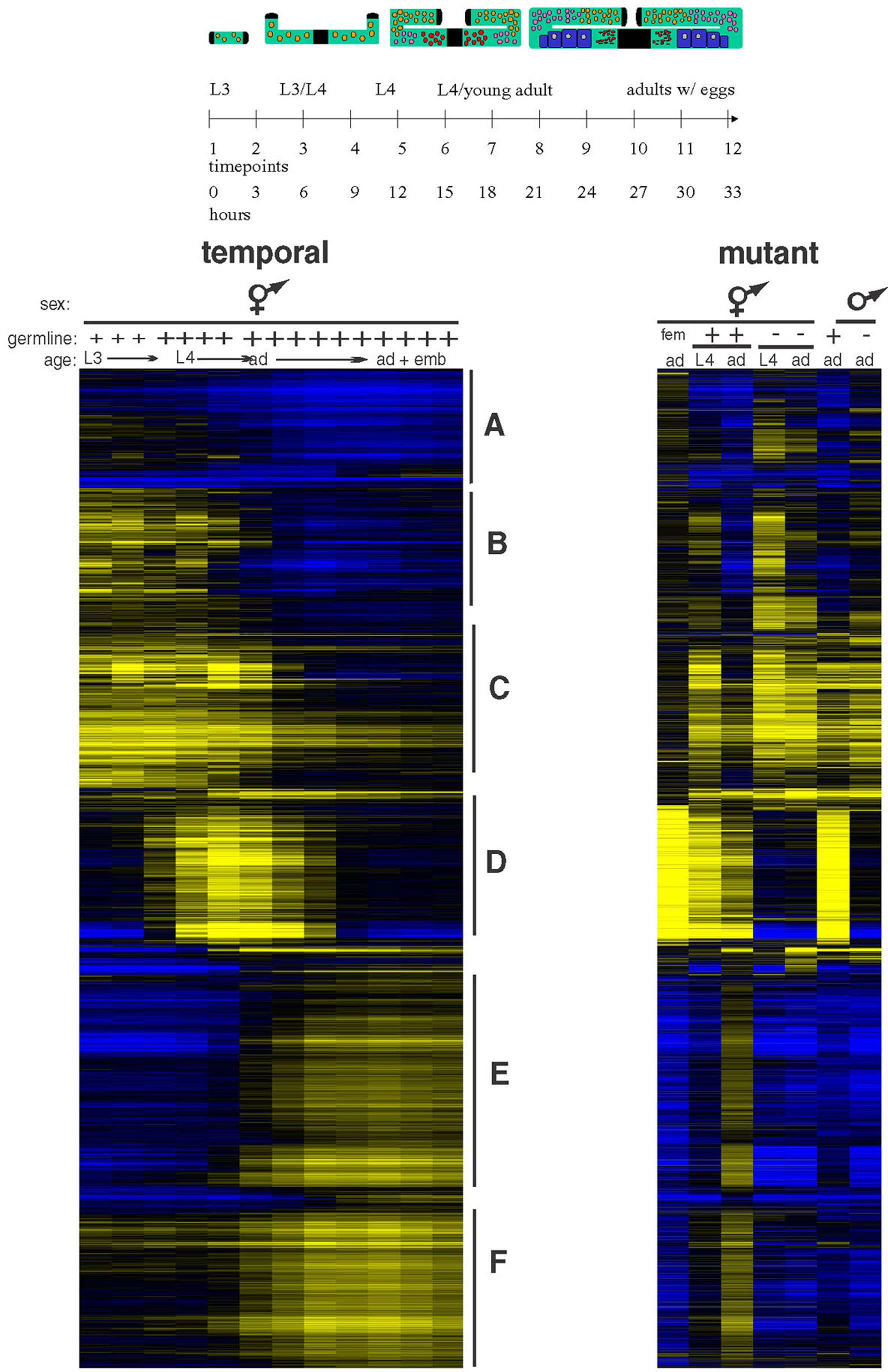
To identify sets of co-regulated genes, gene expression data can be grouped together by similarity of expression profiles. One method for grouping genes is hierarchical clustering using the Pearson correlation coefficient to determine relatedness of expression (Eisen et al., 1998). In Figure 3, hierarchical clustering was performed on the data from the timecourse microarrays. Not surprisingly, some of these clusters based on common temporal regulation contain particular classes of germline genes, as defined by the analysis of mutants (Clusters D-F in Figure 3).
|
Figure 3. Analysis of gene expression in larvae and adults . The top diagram shows germline development during the various stages that were examined. Black = somatic gonad; orange = proliferating germ cells; pink = meiotic germ cells; red = differentiating spermatocytes; blue = differentiating oocytes. The clustergrams below show all genes whose expression changed over the timecourse examined. Each row represents a gene, and, in the clustergram on the left, each column represents a different timepoint in the analysis. The clustergram on the right shows the expression levels of the same genes (in the same order) in wild type, glp-4(ts), fem-1(lf), and fem-3(gf) hermaphrodites, as well as in wild type and glp-4(ts) males. Above the clustergrams, small and large "+" indicate small and large germ line, "–" indicates diminished germ line in glp-4(ts) mutants, and “fem” indicates the fem-3(lf) vs fem-3(gf) experiment. Yellow indicates higher expression in staged wild type, glp-4(ts), or fem-3(gf) samples; blue indicates higher expression in fem-1(lf), or a reference sample that was used for the timecourse. Clusters of genes with distinct temporal expression profiles are marked by letters. Cluster D contains almost exclusively spermatogenesis genes, while intrinsic and oogenesis genes are distributed among clusters E and F. Clusters A-C primarily contain somatic genes whose expression varies over time. Reprinted with permission from Reinke et al., 2004.
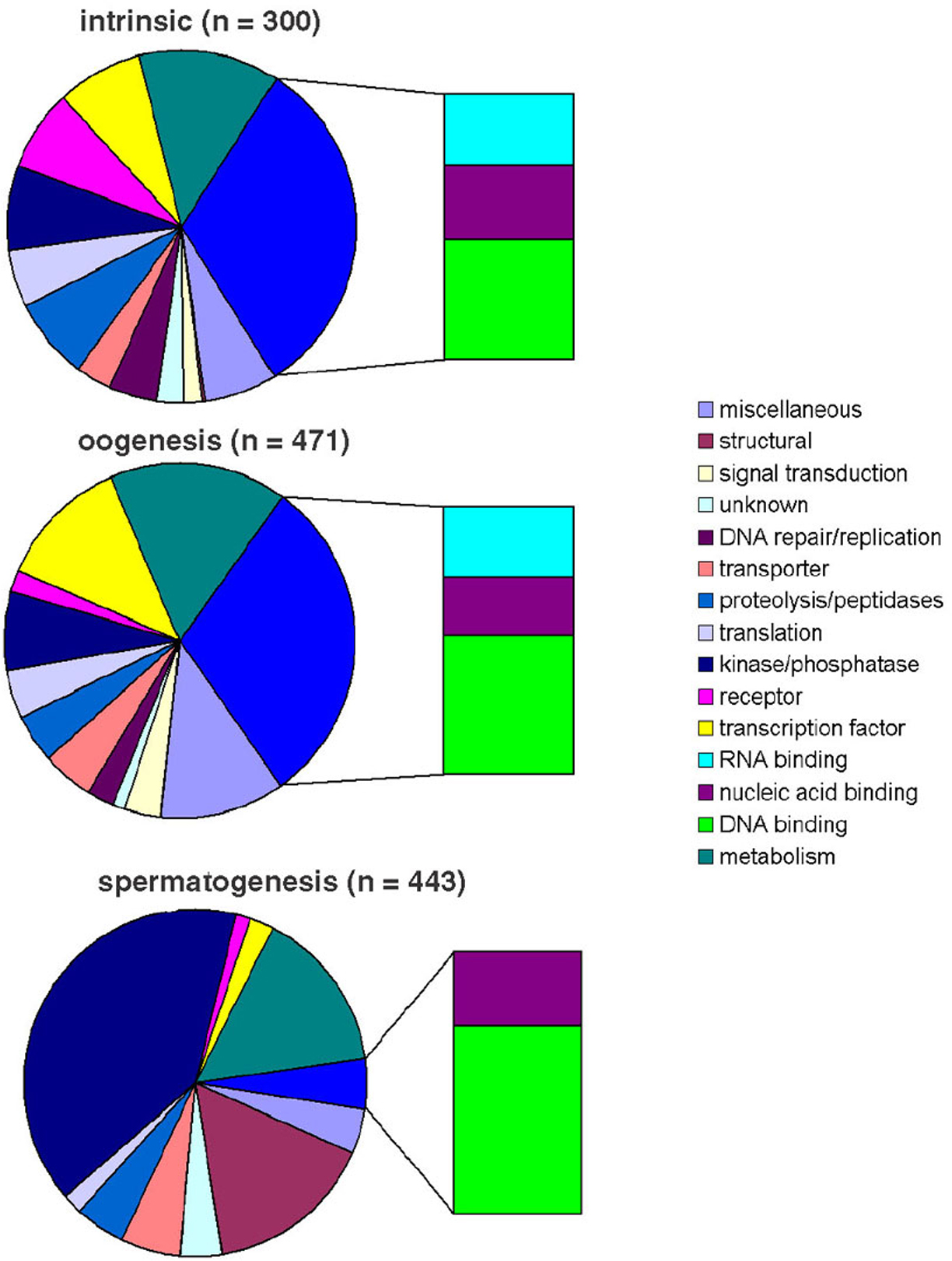
One prominent cluster identified from the timecourse microarrays contains a set of genes with sharp onset of expression in late L3s and abrupt decline in late L4 (cluster D in the temporal clustergram at left in Figure 3). Examination of this set of genes in the microarray analysis performed on mutants (right side of Figure 3) showed that 99% are associated with spermatogenesis. Thus, the spermatogenesis genes are regulated in a fashion that is distinct from the oogenesis and intrinsic genes, and the timing of their expression coincides with the duration of spermatogenesis in late L3/L4 larvae. Additionally, spermatogenesis genes encode different classes of proteins, with more signaling molecules and fewer DNA and RNA metabolism factors (Figure 4). Thus, genes with spermatogenesis-enriched expression are clearly distinct from other germline-enriched genes.
|
Figure 4. Classification of germline-enriched gene sets based on predicted functions. Using Gene Ontology categories, the three sets of germline-enriched genes (spermatogenesis, oogenesis, and intrinsic), were categorized by predicted function of their encoded protein products. The oogenesis and intrinsic groups are composed of very similar proportions of similar types of proteins, while the spermatogenesis group is distinct. The bars represent the different classifications of nucleic acid binding proteins. Reprinted with permission from Reinke et al., 2004.
Unlike the spermatogenesis-enriched genes, expression of other germline-enriched genes (in clusters E and F) increased over time without any significant decline. Overall, the temporal regulation of these germline genes was very similar, but minor differences sub-divided them into two groups. Expression of cluster F genes gradually increases in L3 or L4, while expression of cluster E genes has a more abrupt onset at adulthood. The cluster E expression profile is consistent with oogenesis, which initiates in the adult. However, genes in the intrinsic and oogenesis sets are fairly evenly distributed between cluster E and cluster F. Furthermore, the classes of protein functions are also proportioned similarly in the two clusters (Figure 4). Thus, separation of germline genes into “intrinsic” and “oogenesis” categories is not supported by the temporal analysis, and it is difficult at this point to separate genes specifically required in immature germ cells versus maturing oocytes (see Reinke et al., 2004 for discussion of possible reasons).
Genome-scale views can provide much more than gene lists. A prime example is the discovery of a chromosomal bias in the genomic location of germline-enriched genes, which arose from the microarray studies described above. The germline genes are primarily located on the autosomes; very few reside on the X chromosome (Reinke et al., 2000; Reinke et al., 2004). This bias is particularly strong for spermatogenesis genes, and though less marked for other germline genes, the bias is quite significant.
The biased location of germline-enriched genes, in combination with electron microscopic images of germ cell nuclei in males, led to the hypothesis that the X chromosome might be transcriptionally silent in the germ line. By EM analysis of male germlines, the unpaired X chromosome appears condensed (Goldstein, 1992), a morphology now thought to be similar to the condensed, transcriptionally inactive sex chromosome body observed during meiosis in organisms with single or non-equivalent sex chromosomes (McKee and Handel, 1993). If the X chromosome is essentially transcriptionally inactive in the male germline, then it would be an inhospitable environment for genes needed in that tissue. Thus, genes expressed in the germline would ultimately be under-represented on the X chromosome. Alternatively, some unknown force could prevent germline genes from residing on the X, and consequently, their absence from the X triggers its apparent transcriptional silencing.
Subsequent experiments have supported and extended the hypothesis that the X is silenced in the germline. In the male germline, the unpaired X chromosome is decorated with histone modifications that correlate with transcriptional silencing, and is devoid of modifications that correlate with active gene expression (Kelly et al., 2002; see Germline chromatin). Unexpectedly, the synapsed X chromosomes in hermaphrodites are also apparently silenced throughout most of the germ line, as they largely lack detectable activating modifications (Kelly et al., 2002; see ). However, silencing modifications are not seen on the hermaphrodite Xs as they are in the male (Bean et al., 2004; see Germline chromatin). This milder silencing correlates well with the extent of gene bias against the X chromosome, which is less severe for genes expressed primarily in the hermaphrodite germline (oogenesis genes) compared to those shared with the male germline (spermatogenesis genes). The silencing of the Xs in hermaphrodites was subsequently shown to require the activity of the MES proteins (Fong et al., 2002). Mutations in the mes-2, mes-3, and mes-6 genes causes the X to inappropriately acquire marks of active chromatin, suggesting that transcriptional control of the X chromosome is disrupted (Fong et al., 2002; see Specification of the germ line).
The remarkable insight that an entire chromosome is silenced in a major tissue such as the germ line would have been difficult to achieve if the germ line had been studied only on a gene-by-gene basis. As each individual germline-enriched gene was identified, its location on an autosome would not have been surprising. By examining the expression profiles of many germline genes in parallel, the bias against the X chromosome became obvious, and could be further examined through additional experiments. This example provides an important instance of how emergent properties become apparent from comprehensive molecular depictions.
In addition to the microarray approach described above, other large-scale methods of examining gene expression have been undertaken. An in situ hybridization project performed under the direction of Dr. Yuji Kohara examined the expression pattern of several thousand genes at multiple stages of development, from embryos to young adults, specifically in hermaphrodites. Because in situ hybridization analysis reveals both location and timing of transcript accumulation, it complements the quantitative data provided by microarrays. Together, these two methods provide insights into spatial, temporal, and condition-dependent control of gene expression. The whole-mount in situ studies have successfully identified many genes with germline-enriched expression, perhaps because the germ line has a relatively high rate of mRNA production and/or because the germ line is one of the largest organs in the adult. The spatial aspect of in situ hybridization data is particularly informative. For instance, accumulation of oogenesis transcripts from X-linked germline genes is restricted to the proximal germ line, consistent with silencing of the X chromosome in the distal germ line (Kelly et al., 2002, see Germline chromatin).
The identification of germline-expressed genes provides an excellent starting point for analysis of gene function during oogenesis and early embryogenesis. Indeed such studies have begun. Based on an early version of the germline gene set (Reinke et al., 2000), several RNAi screens have been performed, including ones focused on identification of genes required for early embryogenesis (Piano et al., 2002), genes required for correct localization of PIE-1 in the early embryo (Pellettieri et al., 2003; see Specification of the germline), and genes required for chromosome morphogenesis during meiosis (Colaiácovo et al., 2003). A separate study identifying genes expressed during meiosis was followed by RNAi analysis of candidate genes for roles in germline development (Hanazawa et al., 2001). Additionally, protein partners are being identified by systematic yeast two-hybrid analysis (Walhout et al., 2002). Therefore, a tremendous data set is emerging for germline genes, which includes their temporal and spatial expression, biological functions, and protein partners. As this data set becomes more comprehensive, the data will provide a remarkably complete overview of the function and interaction of critical factors and can be used to assemble the genetic regulatory networks underlying germline and embryonic development.
Many questions about germline development and function remain that will benefit from genomic analyses. Below, some questions are listed along with approaches for answering those questions using DNA microarrays.
How does gene expression differ between different stages of germ cell development? Many additional mutants exist that can be used to further distinguish genes that are expressed in different germ cell types. For instance, a gain-of-function mutation in the GLP-1(Notch) receptor causes excess germline stem cells (Berry et al., 1997; see Germline proliferation and its control). Use of this mutant in microarray experiments will identify genes expressed primarily in the distal proliferating stem cells and better separate these genes from those that are expressed and act in oogenesis.
How many germline-expressed genes still remain to be identified? Because the current microarray studies used intact animals, somatic gene expression has likely masked the germline expression of certain genes. In the future, the use of dissected gonads in microarray experiments will greatly improve the sensitivity with which germline-expressed genes can be identified. Dissected gonads are composed almost entirely of germ cells; with careful dissection, the only somatic cells included are the distal tip cell and sheath cells. Improvements to linear RNA amplification techniques permit the use of very small amounts of RNA (Baugh et al., 2001), and it has been possible to perform microarray experiments with RNA derived from ~50 dissected adult gonads. Use of dissected gonads will also improve detection of gene expression changes in mutants with only subtle germline defects.
How do post-transcriptional mechanisms control target mRNAs? Current germline microarray analyses have identified many RNA-binding proteins that are likely to act in the germ line to regulate mRNA translation and/or stability. The RNA targets bound by these proteins can be identified using microarrays. In this experiment, an antibody specific to the RNA binding protein is used to immunoprecipitate the protein and its associated RNAs. These RNAs can then be isolated, labeled and hybridized to a microarray. This approach has worked well for one germline RNA binding protein, GLD-1 (see RNA-binding proteins; M.H. Lee, V. Reinke, and T. Schedl, unpublished data). Over 100 candidate GLD-1 mRNA targets have been identified, the vast majority of which have germline-enriched expression.
How do DNA- and chromatin-binding proteins affect germline gene expression? The initial germline microarray experiments described above have identified many candidate transcriptional regulators that likely function in the germ line. RNAi or a genetic deletion of the transcriptional regulator will presumably result in changes in the levels of its transcriptional targets that can be assessed by DNA microarray analysis. These expression data can be correlated with the binding locations of these proteins in the genome. Approaches such as chromatin immunoprecipitation (ChIP) or DNA adenine methylase identification (DamID), which identify or mark DNA sequences bound by specific transcriptional regulators, can be combined with DNA microarray technology to map the bound DNA sequences genome-wide (Pollack and Iyer, 2002; van Steensel and Henikoff, 2003). Because most DNA-binding proteins bind in non-coding, regulatory sequences that flank genes, a microarray that contains intergenic sequences is required for this analysis. Such arrays are currently being built by Jason Lieb at the University of North Carolina at Chapel Hill.
Global genome studies will help to further determine the gene regulatory networks that govern germline function. In addition to microarray-based approaches, systematic RNAi analysis of germline-expressed genes will shed significant light on their roles in germline development (see for example Colaiácovo et al., 2003). Proteomic approaches, such as systematic yeast two-hybrid, mass spectrometry analysis of TAP-immunoprecipitated proteins, and systematic structural analysis (for review, see Zhu et al., 2003), will also play important roles in deciphering the functional relationships among germline gene products. When data from these different functional approaches are combined for germline-expressed genes, we will be well-launched on a comprehensive understanding of germline function.
http://wormgermline.yale.edu (data generated in mutant and timecourse microarray studies can be searched and downloaded). http://nematode.lab.nig.ac.jp (Kohara lab in situ database “NExtDB”) http://www.RNAi.org (database of RNAi experiments including germline gene set) http://www.wormbase.org (database of the genomic sequence and useful tools for database mining).
Barton, M.K., Schedl, T., and Kimble, J., (1987). Gain-of-function mutations of fem-3, a sex-determination gene in Caenorhabditis elegans. Genetics 115, 107–119. Abstract
Baugh, L.R., Hill, A.A., Brown, E.L., and Hunter, C.P., (2001). Quantitative analysis of mRNA amplification by in vitro transcription. Nuc. Acids Res. 29, E29. Abstract Article
Bean, C.J., Schaner, C.E., and Kelly, W.G., (2004). Meiotic pairing and imprinted X chromatin assembly in Caenorhabditis elegans. Nat Genet. 36, 100–105. Abstract Article
Beanan, M.J., and Strome, S., (1992). Characterization of a germ-line proliferation mutation in C. elegans. Development 116, 755–766. Abstract
Berry, L.W., Westlund, B., Schedl, T., (1997). Germ-line tumor formation caused by activation of glp-1, a Caenorhabditis elegans member of the Notch family of receptors. Development 124, 925–936. Abstract
Colaiácovo, M.P., Stanfield, G.M., Reddy, K.C., Reinke, V., Kim, S.K., and Villeneuve, A.M., (2003). A targeted RNAi screen for genes involved in chromosome morphogenesis and nuclear organization in the Caenorhabditis elegans germline. Genetics 162, 113–128. Abstract
Duggan, D.J., Bittner, M., Chen, Y., Meltzer, P., and Trent, J.M., (1999). Expression profiling using cDNA microarrays. Nat. Genet. 21, 10–14. Abstract Article
Eisen, M.B., Spellman, P.T., Brown, P.O., and Botstein, D., (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95, 14863–14868. Abstract Article
Fong, Y., Bender, L., Wang, W., and Strome, S., (2002). Regulation of the different chromatin states of autosomes and X chromosomes in the germ line of C. elegans. Science 296, 2235–2238. Abstract Article
Goldstein, P., (1982). The synaptonemal complexes of Caenorhabditis elegans: pachytene karyotype analysis of male and hermaphrodite wild type and him mutants. Chromosoma 86, 577–593. Article
Hanazawa, M., Mochii, M., Ueno, N., Kohara, Y., and Iino Y., (2001). Use of cDNA subtraction and RNA interference screens in combination reveals genes required for germ-line development in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 98, 8686–8691. Abstract Article
Jiang, M., Ryu, J., Kiraly, M., Duke, K., Reinke, V., and Kim, S.K., (2001). Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 98, 218–223. Abstract Article
Kelly, W.G., Schaner, C.E., Dernburg, A.F., Lee, M.H., Kim, S.K., Villeneuve, A.M. and Reinke, V., (2002). X chromosome silencing in the germ line of C. elegans. Development 129, 479–492. Abstract
McKee, B.D. and Handel, M.A., (1993). Sex chromosomes, recombination, and chromatin conformation. Chromosoma 102, 71–80. Abstract Article
Nelson, G.A., Lew, K.K., and Ward, S., (1978). Intersex, a temperature-sensitive mutant of the nematode, C. elegans. Dev. Biol. 66, 386–409. Abstract Article
Pellettieri, J., Reinke,V., Kim, S.K., and Seydoux, G., (2003). Coordinate activation of maternal protein degradation during the egg-to-embryo transition in C. elegans. Dev. Cell 5, 451–462. Abstract Article
Piano, F., Schetter, A.J., Morton, D.G., Gunsalus, K.C., Reinke, V., Kim, S.K., and Kemphues, K.J. (2002). Gene clustering based on RNAi phenotypes of ovary-enriched genes in C. elegans. Curr. Biol. 12, 1959–1964. Abstract Article
Pollack, J.R., and Iyer, V.R., (2002). Characterizing the physical genome. Nat. Genet. 32, 515–521. Abstract Article
Reinke, V., San Gil, I., Ward, S., and Kazmer, K., (2004). Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 131, 311–323. Abstract Article
Reinke, V., Smith, H.E., Nance, J., Wang, J., Van Doren, C., Begley, R., Jones, S.J., Davis, E.B., Scherer, S., Ward, S., and Kim, S.K., (2000). A global profile of germline gene expression in C. elegans. Mol. Cell 6, 605–616. Abstract Article
Van Steensel, B., and Henikoff, S., (2003). Epigenomic profiling using microarrays. Biotechniques 35, 346–357. Abstract
Walhout, A.J.M., Reboul, J., Shtanko, O., Bertin, N., Vaglio, P., Ge, H., Lee, H., Doucette-Stamm, L., Gunsalus, K.C., Schetter, A.G., Morton, D., Kemphues, K.J., Reinke, V., Kim, S.K., Piano, F., and Vidal, M., (2002). Integrating Interactome, Phenome, and Transcriptome Mapping Data for the C. elegans Germline. Curr. Biol. 12, 1952–1958. Abstract Article
*Edited by Judith Kimble and Susan Strome. Last revised March 18, 2005. Published January 20, 2006. This chapter should be cited as: Reinke, V. Germline genomics (January 20, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.74.1, http://www.wormbook.org.
Copyright: © 2006 Valerie Reinke. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: valerie.reinke@yale.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.