Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Germline apoptosis shares with somatic apoptosis a reliance on key components of the core apoptotic machinery, including CED-3 and CED-4. However, germline apoptosis differs from somatic apoptosis in its regulation. Whereas somatic apoptosis is developmentally programmed by cell lineage, germline apoptosis occurs as part of an oogenesis program. One category of germline apoptosis, dubbed “physiological” germline apoptosis, reduces the number of cells that complete oogenesis, and is independent of the BH3-only apoptosis effecter EGL-1. A second category, termed “stress-induced” germline apoptosis, is triggered by a genomic integrity checkpoint. Some mechanisms that are monitored by this DNA-damage checkpoint are also involved in germ cell “immortality,” or preservation of a continuous germ cell lineage over successive generations. In addition, exposure to certain environmental insults or pathogens induces germ cell apoptosis. Here we will review the mechanisms that control each of the pathways leading to germ cell apoptosis and discuss their functional significance. Germline apoptosis is an integral part of oogenesis in many animals, including humans. Because many of the regulators of C. elegans germline apoptosis are conserved, we suggest that this nematode provides a valuable model for understanding controls of germline apoptosis more broadly.
In C. elegans germ cells are a unique tissue in three important ways. As is true in all metazoa, C. elegans germ cells are both pluripotent and immortal, in that they give rise to all cell types in the next generation, and also produce all subsequent generations. Secondly, the germline is the only adult tissue that is maintained by stem cells that constantly replenish its population, as maturing gametes are either used or destroyed. The C. elegans germline is therefore more similar to self-renewing mammalian tissues than to the post-mitotic somatic tissues of C. elegans adults. Finally, the C. elegans germline is the only tissue in which apoptosis is not controlled by an invariant cell lineage. Instead, germline apoptosis appears to be an intrinsic part of the oogenesis program. One type of germline apoptosis involves the death of many early germ cells during normal oogenesis, a remarkable but little-understood phenomenon that seems to be broadly conserved. A second category is triggered by DNA damage or various environmental stresses, a property typical of essentially all cell types in vertebrates. Germline apoptosis can also be triggered by infection with certain pathogenic bacteria. Therefore, the C. elegans germline provides a valuable system for tackling the elusive question of how and why so many early oogenic germ cells die in diverse species, and for analyzing mechanisms that trigger apoptosis during normal oogenesis or in response to stress.
In this chapter, we summarize methods to visualize germline apoptosis and then discuss the two major categories of germline apoptosis. We first review “physiological” germ cell apoptosis, which occurs during normal C. elegans oogenesis (Gumienny et al., 1999). This category of apoptosis is regulated differently from other C. elegans apoptosis, by a pathway that has yet to be identified. We next review germline apoptosis that is induced in response to a genome integrity checkpoint, a process we will refer to as DNA damage-induced germ cell apoptosis (Gartner et al., 2000). This pathway may play an important protective role with respect to maintenance of germ cell function. We also discuss the regulation and possible functions of germline apoptosis induced by other environmental stresses or pathogen exposure (Aballay and Ausubel, 2001; Salinas et al., 2006). We will not cover necrotic germ cell death, which occurs under some abnormal conditions but has not been characterized mechanistically. Finally, we review our current understanding of mechanisms that maintain immortality of the germline over multiple generations.
Several methods have been employed to monitor germ cell apoptosis. This section reviews the advantages and disadvantages of common methods. For a more comprehensive discussion, we refer the reader to the WormBook chapter “Methods in cell biology” and (Gartner et al., 2004). Apoptotic germ cells can be detected by the TUNEL (TdT-mediated dUTP Nick End Labeling) assay, or by the high degree of DNA condensation that is characteristic of apoptotic cells in general; however, both techniques require fixation and may be less sensitive than other methods. The most straightforward method to score germ cell apoptosis is to identify cell corpses based on their distinct morphology, as seen using DIC microscopy (Gumienny et al., 1999). It is also possible to detect apoptotic cells by staining living animals with dyes that detect nucleic acids within apoptotic cells, such as acridine orange (AO) or SYTO 12. These stains have been tremendously useful for quantitating the steady-state numbers of apoptotic germ cells, with the caveat that AO does not work in engulfment-defective mutants.
A sensitive method for visualizing germ cell apoptosis relies on CED-1::GFP, a fusion protein that highlights the somatic sheath cell that clusters around each apoptotic germ cell during engulfment (Schumacher et al., 2005). CED-1::GFP marks only early apoptotic corpses, which often cannot be discerned by DIC optics. This method is highly sensitive and convenient, but is not suitable when large numbers of cell deaths occur: CED-1::GFP then appears diluted among a large number of dying cells, making detection of individual cells difficult. In addition, time lapse analyses indicate that some cells surrounded by CED-1::GFP may not end up forming apoptotic corpses (Aymeric Bailly and Anton Gartner, unpublished).
The number of apoptotic germ cells detected in the gonad at any given time can be affected by additional variables besides the proportion of germ cells that undergo apoptosis. For example, the steady-state number of apoptotic cells will be influenced by changes in the rate of corpse engulfment. When analyzing germ cell apoptosis, it is therefore important to investigate whether a particular stimulus or mutant alters the frequency of apoptosis independently of any possible effects on engulfment. This issue can be addressed by using DIC microscopy to count the number of germ cell corpses that accumulate in the engulfment-defective ced-1 background (Gumienny et al., 1999). Alternatively, by performing time lapse analysis, it is possible to monitor the kinetics of apoptotic germ cell corpse formation and removal directly. In addition, the rate at which corpses are generated is theoretically determined not only by the probability that oogenic germ cells will die, but also by the rate at which germ cells are produced. It is therefore important to examine germ cell proliferation in parallel to apoptosis. Common methods to assess proliferation include counting total germ cell number, analysis of the mitotic index or determining the rate of egg laying (Gartner et al., 2004; Methods in cell biology). In general, given that multiple variables can influence germ cell apoptosis, it is impossible to discriminate among small differences in germ cell apoptosis frequency, as can be done for somatic apoptotic death during development (Schwartz, 2007).
|
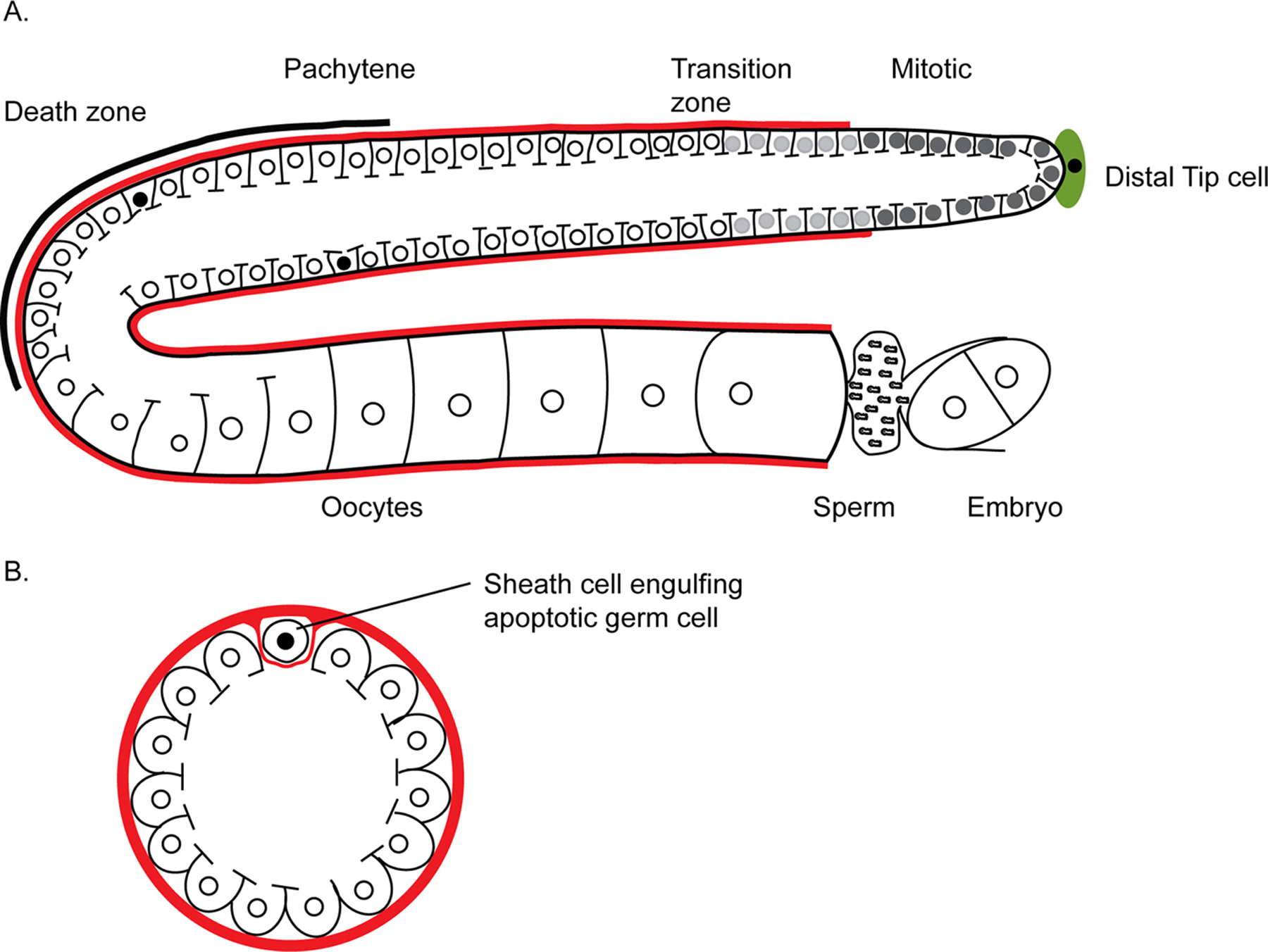
Figure 1. The C. elegans hermaphrodite gonad, and germline apoptosis. (A) Schematic representation of an adult hermaphrodite gonad. One of two gonad arms is shown. The basic features of C. elegans germ cell production have been described elsewhere (see Introduction to the germline, and Germline proliferation and its control). Briefly, germ cells are derived from a proliferating stem cell population located at the distal end of the gonad (relative to the uterus). These germline stem cells give rise to sperm during larval stages, then switch to production of oocytes during adulthood. The somatic distal tip cell regulates germline stem cell proliferation, and entry into the transition zone and prophase of Meiosis I. Germ cells are partially enclosed by a plasma membrane, and share a common cytoplasmic core. Germ cell apoptosis occurs mainly within the gonad loop region (black line), approximately as developing oocytes exit the pachytene stage of meiotic prophase. Apoptotic nuclei are shown as solid black. Apoptosis of germ cells occurs during oogenesis, and therefore is not seen in larvae or males. After pachytene exit, the surviving germ cells grow rapidly and differentiate into individual cellularized oocytes in the proximal germline, and are fertilized in the spermatheca after they undergo maturation (see Control of oocyte meiotic maturation and fertilization). (B) Cross section through the loop region, showing a single cell undergoing apoptosis. Each dying cell rapidly “pinches off” from the shared cytoplasmic core and forms a discrete cell that contains very little cytoplasm. The apoptotic cell is engulfed and removed by one of the somatic sheath cells that surround the gonad (Gumienny et al., 1999).
The landmark study of (Gumienny et al., 1999) first analyzed C. elegans germline apoptosis and revealed that this cell death occurs frequently under normal conditions. In C. elegans, germ cell apoptosis occurs only during oocyte production, and therefore is seen only in adult hermaphrodites. Germline apoptosis is further restricted to the gonadal loop region, where developing oocytes exit the pachytene stage of meiotic prophase and transition into diplotene (Figure 1A). Most germ “cells” are only partially enclosed by a plasma membrane, and connect with each other via cytoplasmic bridges opening onto a common cytoplasm called the rachis (Figure 1A). When a germ “cell” dies, the doomed nucleus initially becomes cellularized to form a discrete cell that contains very little cytoplasm; most of its surrounding cytoplasm is left behind in the rachis (Gumienny et al., 1999). The dying cell then undergoes apoptosis to produce a corpse that is engulfed by neighboring somatic sheath cells (Figure 1B). The engulfment mechanism for germline corpses is the same as for somatic corpses.
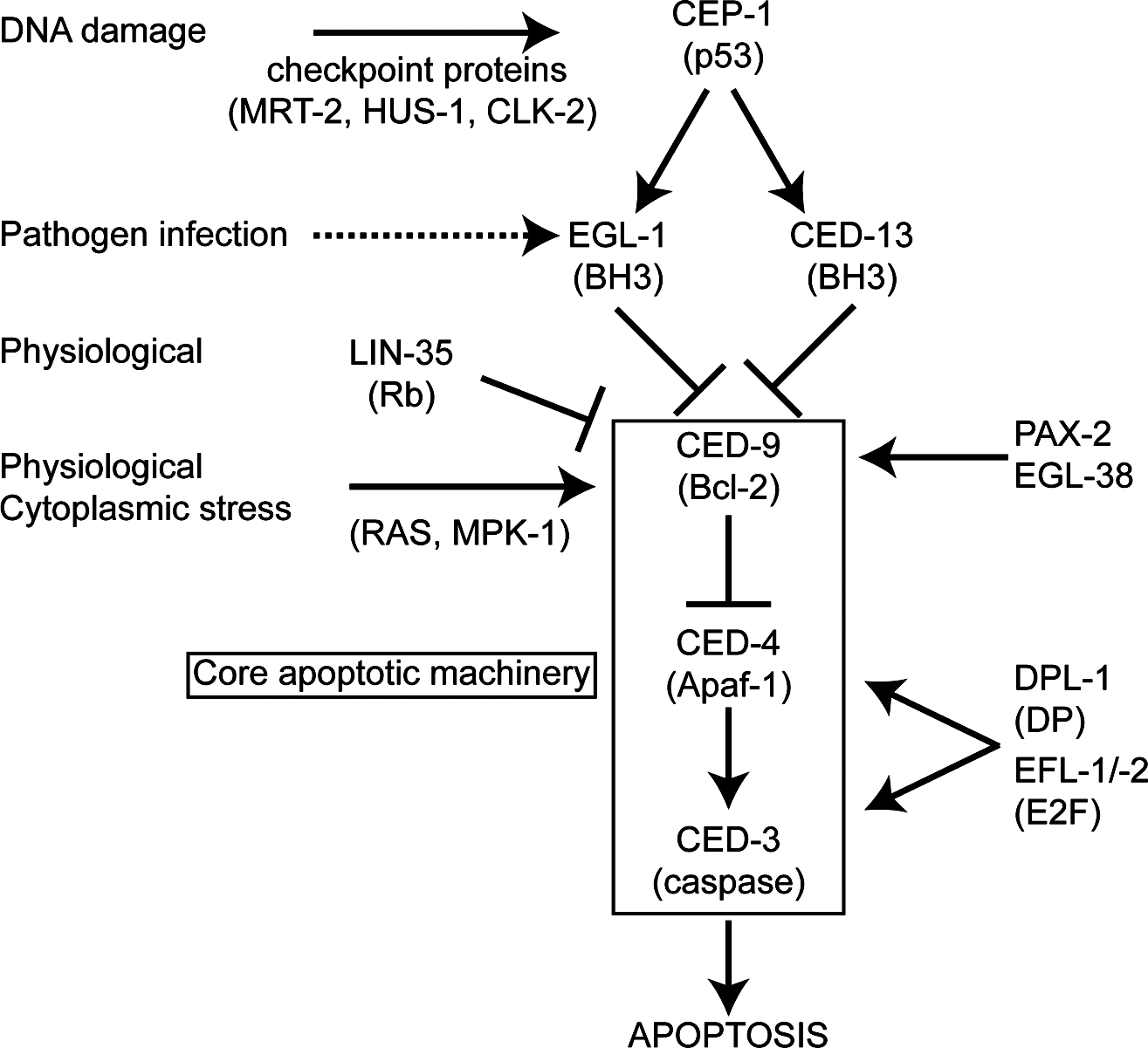
Physiological germline apoptosis occurs in the absence of any known stress and is initiated by the core apoptotic machinery, including CED-3 and CED-4 (Figure 2; see Programmed cell death; Lettre and Hengartner, 2006). However, its induction does not rely on the pro-apoptotic BH3-only protein EGL-1, and therefore differs from other C. elegans apoptosis (Figure 2). Moreover, physiological germ cell apoptosis is not prevented by a gain-of-function mutation (n1950) of the death-inhibitory protein CED-9 (Gumienny et al., 1999). Therefore, physiological germ cell death involves induction of the core apoptosis machinery by an unknown mechanism, making it of significant interest and a topic of current research.
|
Figure 2. Regulation of germ cell apoptosis. At least two distinct pathways of germ cell apoptosis function in C. elegans. Physiological apoptosis is cep-1-independent and a normal feature of oogenesis. The frequency of cep-1-independent germ cell apoptosis increases in response to various cytoplasmic stresses (oxidative, heat, osmotic, starvation). It is not clear whether this increase involves the same triggering mechanism as physiological apoptosis. In each case, these apoptotic events depend upon Ras/MAP-kinase signaling. CEP-1-dependent apoptosis is induced by a DNA damage or chromosomal integrity checkpoint. Rb and E2F transcription factor subunits promote physiological cell death, and appear to function downstream of or in parallel to CEP-1 in the DNA damage response. Pathogen-induced germ cell apoptosis is arbitrarily shown as acting directly on EGL-1, because it has not been determined whether CEP-1 is involved. Physiological and DNA damage germ cell apoptosis pathways are suppressed by the PAX-2 and EGL-38 Pax-family proteins, which increase transcription of the anti-apoptotic protein CED-9.
Physiological germ cell apoptosis commences in early adulthood, increases over time, and is thought to eliminate at least half of all oogenic germ cells that are generated (Gumienny et al., 1999). Physiological apoptosis is not required for oogenesis, but if this cell death is blocked, the number of offspring is reduced under some conditions (Boag et al., 2005; Gumienny et al., 1999). Why might it be advantageous for so many developing oocytes to die? The most attractive model involves the idea that germ cells in pachytene serve as nurse cells for maturing oocytes. According to this model, an excess of germ cells supply cytoplasmic components essential for growth of developing oocytes, and a stochastic selective mechanism culls the number of germ cells allowed to differentiate into oocytes, thereby increasing the amount of cytoplasm available to each enlarging oocyte (Gumienny et al., 1999).
Considerable evidence supports the “nurse cell” model. Physiological germ cell apoptosis occurs in the region of the gonad where developing oocytes begin to enlarge (Figure 1A), thus maximizing the time that these nuclei survive to serve as nurse cells. Electron microscopy studies indicate that in this region germ cell nuclei all appear generally normal, aside from the small number that have already begun to undergo apoptosis. The doomed nuclei become cellularized rapidly, and are subsequently engulfed in a manner that provides most of their surrounding cytoplasm to the central gonad rachis (Figure 1B; Gumienny et al., 1999). When apoptosis is prevented (in ced-3 or ced-4 mutants), young animals do not produce obviously abnormal oocytes, which would be expected if physiological germ cell apoptosis were culling defective nuclei, but older germlines accumulate oocytes that are smaller than normal. Within the central gonad rachis, cytoplasm actually flows towards the cellularizing oocytes at the proximal gonad end, suggesting that in general pachytene stage germ cells function as “factories” that provide cytoplasmic components to those oocytes (Wolke et al., 2007). Finally, physiological germline apoptosis does not occur during spermatogenesis. Sperm contain very little cytoplasm, making it possible for all sperm nuclei to form gametes without cytoplasm becoming limiting (Gumienny et al., 1999). Sperm precursors also do not undergo apoptosis in response to stress, perhaps because sperm may be more expendable than oocytes.
It is not known how the core cell death mechanism is triggered in physiological germ cell apoptosis, but a number of genes and processes have been identified that affect the frequency of this cell death. For example, Ras/Mitogen-activated protein kinase (MAPK) signaling is a key regulator of physiological germline apoptosis (Gumienny et al., 1999; Park et al., 2006). This apoptosis is prevented if MAPK signaling is blocked, and occurs at excessive levels when MAPK signaling is aberrantly activated. Increased germline apoptosis is seen when MAPK is released from inhibition by loss of either the predicted MAPK phosphatase LIP-1 or the predicted RNA-binding protein GLA-3, which influences various somatic Ras/MAPK-dependent events (Kritikou et al., 2006; Lettre et al., 2004). Ras/MAPK signaling is required for many steps in meiosis, including exit from the pachytene stage (Church et al., 1995; Hajnal and Berset, 2002; Leacock and Reinke, 2006). It is therefore still unclear whether this pathway provides a primary initiating signal for physiological germ cell apoptosis, or directs germ cells to progress to a stage where they are more susceptible to pro-apoptotic signals (Gumienny et al., 1999).
C. elegans contains a single anti-apoptotic Bcl-2 family protein, CED-9, which is essential for inhibiting cell death. An exciting recent study showed that C. elegans Pax transcription factors, which influence apoptosis in many species, prevent inappropriate germ cell and somatic apoptosis by directly activating ced-9 transcription (Figure 2; Park et al., 2006). In addition, physiological germ cell apoptosis occurs in the absence of MAPK signaling in hermaphrodites with decreased CED-9 levels (Gumienny et al., 1999; Park et al., 2006), suggesting that regulation of ced-9 expression may be important for establishing the underlying sensitivity of germ cells to apoptosis. Further supporting this idea, the C. elegans ortholog of the retinoblastoma susceptibility protein RB (LIN-35) is required for physiological germ cell apoptosis to occur at normal levels, because it reduces ced-9 expression (Schertel and Conradt, 2007; Figure 2). In addition, subunits of the E2F transcription factor, which targets RB to genes, seem to promote physiological germ cell apoptosis by increasing the expression of ced-3 and ced-4. RB and E2F components are also required for DNA damage-induced apoptosis to occur. Taken together, these results suggest that regulation of the core apoptotic apparatus at the transcriptional level may be of general importance for modulating germ cell apoptosis.
Additional genes that prevent physiological germ cell apoptosis from occurring at excessive levels have been identified in various studies (Table 1), including a whole genome RNAi screen, and a targeted RNAi screen that tested a large set of genes involved in cell signaling and protein trafficking (Lackner et al., 2005; Lettre et al., 2004). This list is almost certainly incomplete, because RNAi is not always fully penetrant, and because the whole genome RNAi screen specifically identified positives that did not result in sterility (Lettre, et al., 2004). Nevertheless, it is striking that only a modest number of genes have been identified that are required to prevent physiological germ cell apoptosis from occurring at excessive levels (we refer to these as germ cell apoptosis genes). This suggests that only a limited set of cellular functions may affect physiological germ cell apoptosis. Increases in this apoptosis do not in general represent a non-specific response to a catastrophic oogenesis defect, since null or strong loss-of-function mutants in some germ cell apoptosis genes are fertile (car-1, cpb-3, gla-3), and in other cases, RNAi increases apoptosis dramatically without causing sterility (Table 1). When some germ cell apoptosis genes are lacking, fertility is reduced if apoptosis is prevented (Boag et al., 2005), suggesting that the physiological germ cell apoptosis pathway responds to and may ameliorate particular defects in oogenesis.
Table 1. Genes that prevent excessive physiological germ cell apoptosis
| Gene | Protein Class | Reported RNAi or mutant phenotype |
|---|---|---|
| car-11,3 | RGG-box RNA-binding | Fertile, but with extended pachytene region and dead progeny |
| cgh-11,4 | DEAD-box RNA helicase | Oocytes formed, but sterile |
| ced-95 | Bcl2 homolog and general apoptosis inhibitor | Fertile after feeding RNAi |
| cpb-31,3,5 | CPEB ortholog, RNA-binding | Fertile, with extended pachytene region and fewer progeny |
| gla-35,6 | Cys3His Zn finger, RNA-binding | Fertile, with extended pachytene region and fewer progeny |
| gld-17 | KH domain RNA-binding, translation regulator | Weak loss-of-function mutant is fertile with elevated apoptosis |
| lip-16 | MAPK (MPK-1) phosphatase | Accelerated oocyte development and partial embryonic lethality; reduced germline proliferation10 |
| pax-2, egl-388 | Pax proteins, directly induce ced-9 transcription | Elevated somatic and germline apoptosis, developmental defects |
| pmk-35 | p38 MAPK | Fertile after feeding RNAi |
| rab-5, rab-72,9,10 | Ras-related GTPase | Required for engulfment |
| RabGDI/ Y57G11C.102,9 | Rab guanine nucleotide association inhibitors | N.R. |
| RabGGT/M57.2, B0280.12,9 | Rab geranyl geranyl transferase; prenylates Rab proteins | N.R. |
| rfp-15 | RING finger, ubiquitylation | Fertile after feeding RNAi |
| vps-11, vps-16, vps-18, vps-33, vps-39, vps-412,9 | HOPS complex; interacts with Rab7 | vps-41: maternal effect embryonic lethality and partial larval arrest. Otherwise, N.R. |
| Physiological germ cell apoptosis is defined here as being cep-1- and egl-1-independent (see text). 1The proteins encoded by these genes interact with each other physically and functionally in multiple species, and are present in mRNA-protein P-bodies (see text). 2Each of these proteins is involved in endosomal or endosomal-lysosomal membrane fusion and protein trafficking. 3(Boag et al., 2005), 4(Navarro et al., 2001), 5(Lettre et al., 2004), 6(Kritikou et al., 2006), 7(Schumacher et al., 2005), 8(Park et al., 2006), 9(Lackner et al., 2005), 10(Kinchen et al., 2008). C. elegans gene designations for vps-11, vps-16, vps-18, vps-33, and vps-39 can be found in (Lackner et al., 2005). N.R., not reported. | ||
Many germ cell apoptosis genes encode RNA-binding proteins (Table 1). Remarkably, in diverse species three of these proteins (CGH-1, CAR-1, CPB-3) interact physically and functionally with each other, and have been implicated in translational regulation of mRNA that is sequestered in oocytes and other cells (Audhya et al., 2005; Barbee et al., 2006; Boag et al., 2005; Decker and Parker, 2006; Squirrell et al., 2006; Weston and Sommerville, 2006; Wilczynska et al., 2005; Wilhelm et al., 2005). Each of these three proteins is also found in protein-mRNA processing (P)-bodies, cytoplasmic structures in which multiple mRNA translational suppression and degradation mechanisms are congregated, suggesting a link between some P-body functions and physiological germ cell death. In addition, lack of other RNA-binding proteins leads to germ cell apoptosis that has not been established to be egl-1-independent, but might be mediated through the physiological pathway. These RNA-binding proteins include the CPB-3-associated protein DAZ-1 (Hasegawa et al., 2006; Karashima et al., 2000), and the PUF-family proteins FBF-1 and -2 (Cho et al., 2007). Interestingly, germ cell death associated with a depletion of FBF-1 and -2 occurs only in the absence of FSHR-1, the somatic glycoprotein hormone receptor ortholog (Cho et al., 2007). Fertility was restored when this cell death was blocked, further supporting the idea that germ cell apoptosis is regulated by specific signals, and is not simply a non-specific consequence of a developmental catastrophe.
Many proteins that profoundly influence physiological apoptosis are involved in mRNA metabolism. One possibility is that these mRNA regulatory factors affect the expression of apoptosis control proteins. For example, the mRNA-binding protein GLD-1 directly represses translation of cep-1, which is required for DNA damage-induced germ cell apoptosis (see Section 6; Schumacher et al., 2005). GLD-1 also reduces the frequency of cep-1-independent apoptosis, suggesting that it might inhibit translation of an additional pro-apoptotic factor. Alternatively, these RNA binding proteins might influence a “checkpoint” that monitors allocation of cytoplasmic contents as oocytes begin to cellularize. Considering the latter idea, it is interesting that cep-1-independent germ cell apoptosis also appears to increase dramatically when a set of proteins involved in endosomal membrane and protein trafficking is lacking, although it it not yet known whether the increased steady state level of apoptotic corpses seen upon RNAi of these genes is due to excessive apoptosis or delayed corpse engulfment (Table 1). These genes include the C. elegans orthologs of the Rab5 and Rab7 GTPases along with multiple members of the HOPS complex, which interacts functionally with these GTPases. In addition, the death-inhibiting RNA-binding proteins CAR-1 and CGH-1 (Table 1) are required for early embryonic cytokinesis, and CAR-1 and its Drosophila ortholog Trailerhitch have been implicated in endoplasmic reticulum membrane organization and protein trafficking, respectively (Audhya et al., 2005; Boag et al., 2005; Squirrell et al., 2006; Wilhelm et al., 2005). These last observations suggest a third speculative model; perhaps the frequency of germ cell apoptosis is influenced by the processes required for individual oocytes and apoptotic germ cell nuclei to become fully enclosed by the plasma membrane. If apoptosis signals spread inappropriately when apoptotic nuclei are not effectively isolated from their neighbors, this could explain why corpses are frequently seen in clusters when germ cell apoptosis is abnormally elevated (Gartner et al., 2004).
Physiological germ cell apoptosis during C. elegans oogenesis may be conserved broadly among eukaryotes, including humans (Baker, 1963; Borum, 1961; Pepling, 2006). During early meiotic prophase, developing mammalian oocytes are interconnected within cysts and transfer cytoplasmic components amongst themselves. Around or just after the time of birth, approximately two thirds of these developing oocytes die by apoptosis; concomitantly, the cytoplasmic connections break down and the oocytes cellularize (Pepling, 2006; Pepling and Spradling, 2001). The surviving oocytes form primordial follicles. The parallels to C. elegans physiological apoptosis are striking. Mammalian oocyte apoptosis occurs around or just after the transition from pachytene to diplotene, and is associated with the loss of intercellular communication between oocytes (Pepling and Spradling, 2001). In fact, it has been proposed that the eliminated mammalian oocytes act as nurse cells, as in C. elegans (Pepling, 2006). Another intriguing model is that germ cell apoptosis eliminates defective mitochondrial genomes and prevents their transfer to the next generation (Krakauer and Mira, 1999). This is largely theoretical, however, because little is understood about how mitochondrial DNA integrity is maintained over generations in any organism. As mechanisms that regulate C. elegans physiological germ cell apoptosis are identified, it will be of intense interest to investigate their possible role in the developmental apoptosis that occurs during mammalian oogenesis.
The discovery of physiological germ cell apoptosis prompted studies to test the possibility that DNA damage might induce germ cell apoptosis (Gartner et al., 2000). The correct maintenance and duplication of genetic information is constantly challenged by genotoxic stress, which may occur as a consequence of mistakes in DNA replication, misincorporation of nucleotides, oxidative damage, irradiation, or exposure to genotoxic chemicals. In response to genotoxic stress, cells activate conserved DNA damage checkpoint pathways that lead to DNA repair, to transient cell cycle arrest, or to apoptosis and the demise of genetically damaged cells that might harm the organism. Studies focusing on DNA damage responses after ionizing irradiation (IR) revealed the first example of C. elegans apoptosis that was induced by DNA damage (Gartner et al., 2000). That discovery set the stage for genetic dissection of DNA damage response pathways, including both evolutionarily conserved and novel components. It turned out that germ cells, but not somatic cells, are able to undergo DNA damage induced apoptosis. As is the case for physiological apoptosis, only pachytene stage germ cells undergo apoptosis upon stress, and stress-induced apoptosis has not been observed in the male germline. Germ cell apoptosis induced by genotoxic insults is morphologically indistinguishable from physiological germ cell apoptosis, and requires the core apoptotic genes ced-9, ced-4, and ced-3 (Figure 2). However, in contrast to physiological germ cell apoptosis, the worm BH3-only protein EGL-1 is required for DNA damage to induce germ cell apoptosis, indicating that this stress-induced apoptosis is regulated through a different pathway.
The notion that double strand breaks might be the trigger of IR-induced apoptosis is supported by the correlation between elevated levels of double strand breaks and elevated levels of germ cell apoptosis in DNA double strand break repair mutants such as brca-1, brca-2 and rad-51 (Alpi et al., 2003; Boulton et al., 2004; Chin and Villeneuve, 2001; Gartner et al., 2000; Martin et al., 2005; Whetstine et al., 2006). Similarly, RNAi of the histone H3 K9/K36 histone demethylase JMJD2A leads to elevations in double strand DNA breaks and germ cell apoptosis (Whetstine et al., 2006). In addition, more recent studies have implicated multiple additional genotoxic insults such as UV irradiation (Derry et al., 2007; Stergiou et al., 2007), DNA alkylating agents (Holway et al., 2006), and misincorporation of uracil into DNA as triggers for germ cell apoptosis (Dengg et al., 2006). Some of these DNA damaging agents do not necessarily cause increased levels of DNA double strand breaks, and their effects can be genetically separated from IR-induced apoptosis (see below).
Multiple genes are required specifically for induction of DNA damage-induced apoptosis, and not for somatic cell apoptosis or physiological germ cell apoptosis (Gartner et al., 2000). Amongst mutants that are defective specifically in DNA damage-induced apoptosis, those with defects in upstream DNA damage sensing and signal transduction are predicted to be defective in all DNA damage responses, including DNA repair and transient mitotic germ cell cycle arrest. Sensitivity to DNA damage is scored via the so-called Rad survival assay, where worms are irradiated at the L4 larval stage, and levels of irreparable DNA damage in affected germ cells are assessed by scoring the survival rates of the resulting progeny (Gartner et al., 2004). The first mutations associated with defects in irradiation-induced apoptosis turned out to impair all DNA damage responses, and were later mapped to the mrt-2, clk-2 and hus-1 loci (Table 2). mrt-2 and hus-1, which also function in telomere replication (see Section 9), have homologs in mammals and yeasts (Ahmed and Hodgkin, 2000; Gartner et al., 2000; Hofmann et al., 2002). These proteins are part of the conserved 9-1-1 PCNA-like DNA sliding complex, which associates with DNA double strand breaks in vivo. Loading of the 9-1-1 complex depends on the HPR-17 clamp loader, which has a role in telomere elongation and is functionally conserved in C. elegans (Table 2; Boerckel et al., 2007).
Table 2. Genes implicated in germline DNA damage responses
| Gene | Protein class | Notes | Essential for viability | IR induced apoptosis | IR cell cycle arrest | IR sensitivity (repair of IR ind. DNA damage) | UV induced apoptosis | UV cell cycle arrest | S-phase check- point |
|---|---|---|---|---|---|---|---|---|---|
| atm-11 | PI3 kinase, homolog of ATM | conflicting data | no | +/− | +/− | nd | + | + | + |
| atl-12 | PI3 Kinase homolog of ATR | yes | – | – | nd | – | – | – | |
| clk-23 | conserved DNA damage checkpoint protein | yes | – | – | +/− | – | – | – | |
| hpr-174 | homolog of S. pombe/human Rad-17p clamp loader | no | – | – | – | nd | nd | +/− | |
| mrt-25 | homolog of S. pombe/human Rad-1p 9-1-1 sliding clamp component | no | – | – | – | +/− | +/− | +/− | |
| hus-16 | homolog of S. pombe/human Hus-1p 9-1-1 sliding clamp component | no | – | – | – | +/− | +/− | +/− | |
| hpr-97 | homolog of S. pombe/human Rad-9p 9-1-1 sliding clamp component | RNAi only | no | – | – | nd | nd | nd | +/− |
| cep-18 | Primordial p53 or p63 family member | no | – | + | – | – | – | + | |
| chk-19 | homolog of human Chk-1 protein kinase | yes | nd | – | nd | nd | nd | – | |
| chk-210 | homolog of human Chk-2 protein kinase | yes | + | + | nd | – | – | + | |
| xpa-111 | xpa excision repair gene | no | + | + | nd | – | nd | nd | |
| xpg-111 | xpg excision repair gene | no | + | + | nd | +/− | nd | nd | |
| phg-112 | related to GAS growth arrest related | RNAi only | no | + | + | + | – | – | – |
| sex-113 | nuclear hormone receptor like transcription factor, X-chromosome signal element | RNAi only | yes | – | +/− | – | nd | nd | nd |
| ina-113 | integrin alpha chain | RNAi only | yes | – | +/− | – | nd | nd | nd |
| F08C6.213 | lipid-activated phosphocholine cytidylyltrans- ferase | RNAi only | nd | – | – | – | nd | nd | nd |
| C25A1.613 | snoRNP complex, subunit | RNAi only | yes? | – | +/− | – | nd | nd | nd |
| F59C6.413 | Exosomal 3′-5′ exoribonuclease complex subunit | RNAi only | yes | – | +/− | – | nd | nd | nd |
| F43D2.113 | Cyclin K | RNAi only | ? | – | +/− | – | nd | nd | nd |
| pdi-214 | Protein disulfate isomerase | RNAi only | yes | +/− | – | nd | nd | nd | nd |
| C04F12.3 ikb-114 | Human bcl-3 like Gene, mutant no phenotype | RNAi only | no | +/− | – | nd | nd | nd | nd |
| sel-8 (lag-3)15 | Notch coactivator | RNAi only | yes | – | nd | nd | nd | nd | nd |
| DNA damage response defects of various mutations and RNAi depletions are listed. nd. not documented. + indicates wild type responses, − indicates defects in DNA damage responses. 1(Garcia-Muse and Boulton, 2005; Stergiou et al., 2007), 2(Garcia-Muse and Boulton, 2005), 3(Ahmed et al., 2001; Stergiou et al., 2007), 4(Boerckel et al., 2007), 5(Gartner et al., 2000; Stergiou et al., 2007), 6(Hofmann et al., 2002; Stergiou et al., 2007), 7(Boulton et al., 2002), 8(Derry et al., 2001; Schumacher et al., 2001), 9(Kalogeropoulos et al., 2004), 10(MacQueen and Villeneuve, 2001; Stergiou et al., 2007; Stergiou et al., 2007), 11(Stergiou et al., 2007), 12(Derry et al., 2007), 13(van Haaften et al., 2006), 14(Boulton et al., 2002), 15(Zhao et al., 2007). | |||||||||
The worm homologs of the mammalian ATM and ATR kinases, which act as upstream DNA damage sensors and are mutated in human genome instability syndromes, have also been implicated in DNA damage checkpoint signaling (Garcia-Muse and Boulton, 2005; Stergiou et al., 2007). Worm ATR is needed for checkpoint responses to IR, UV, and replication stress, and also for an essential function in germ cell development that is most likely linked to DNA replication (Garcia-Muse and Boulton, 2005; Stergiou et al., 2007; Table 2). ATR binds to damaged DNA upon either irradiation or replication stress (Garcia-Muse and Boulton, 2005). ATM is thought to play a minor role in the DNA damage checkpoint response, as atm mutants are only partially defective in responding to IR. However, some conflicting data based on atm RNAi suggest a more prominent role of ATM in IR-induced apoptosis (Garcia-Muse and Boulton, 2005; Stergiou et al., 2007).
The final two conserved kinase components of DNA damage checkpoint signaling are chk-1 and chk-2 (Kalogeropoulos et al., 2004; MacQueen and Villeneuve, 2001). Recent studies implicated chk-1 in mediating IR induced cell cycle arrest and apoptosis (Kalogeropoulos et al., 2004; Table 2). chk-2 is needed for meiotic chromosome pairing and for mediating UV-triggered germ cell apoptosis, but not for IR and replication stress-induced checkpoint responses (MacQueen and Villeneuve, 2001). Interestingly, the nucleotide excision repair genes xpa-1 and xpg-1, which are required for removal of UV-induced DNA lesions such as cyclobutane pyrimidine dimers and 6-4 photoproducts (Costa et al., 2003; Prakash and Prakash, 2000), also seem to have a function in the DNA damage sensing checkpoint, as UV-induced apoptosis is dramatically reduced in these mutants (Table 2; Stergiou et al., 2007).
clk-2 is a functionally conserved checkpoint gene that was first uncovered by C. elegans genetics. The mn159 allele was found in a screen for mutations conferring hypersensitivity to IR, and called rad-5(mn159), while clk-2(qm37) was found in a screen for mutations that slow worm development and lead to an extended life span (Benard et al., 2001; Hartman and Herman, 1982). The clk-2(qm37) lifespan extension is weak, and may derive indirectly from its slow growth phenotype (Harris et al., 2006). Both clk-2 mutations are temperature sensitive, resulting in embryonic lethality at restrictive temperature, while checkpoint defects in response to IR and UV-induced DNA damage and replication stress occur at permissive temperature (Ahmed et al., 2001; Stergiou et al., 2007; Table 2). The CLK-2 protein is the ortholog of S. cerevisiae tel2, which has been implicated in regulation of yeast telomere length and binds telomeric DNA in vitro (Kota and Runge, 1999). In C. elegans clk-2 does not seem to affect telomere length (Ahmed et al., 2001), but has been implicated in sensing misincorporation of uracil into DNA (Dengg et al., 2006). Uracil incorporation results from use of dUTP instead of dTTP by DNA polymerases and is the most common type of endogenous damage. Excessive dUTP incorporation leads to cell cycle arrest and apoptosis in the germline, and ultimately to lethality. The DNA damage response phenotypes can be suppressed by mutations in either clk-2 or the ung-1 base excision repair gene. Therefore, ung-1 may have a DNA damage checkpoint function in addition to its previously known role in base excision repair. In contrast to clk-2, mrt-2 cannot suppress apoptosis and cell cycle arrest triggered by excessive levels of dUTP incorporation.
Recent studies indicate that clk-2 is functionally conserved from yeast to humans. The fission yeast homolog has been implicated in S-phase regulation and DNA damage checkpoint responses (Shikata et al., 2007). Human CLK-2 physically interacts with the ATR complex, and is required for both the replication checkpoint and DNA crosslink repair pathway, where it affects the DNA damage-induced monoubiquitination of the FancD2 repair protein (Collis et al., 2007). Mechanistically, CLK-2 seems to promote S-phase checkpoint activation by preventing CHK-1 kinase degradation (Collis et al., 2007). In fission yeast CLK-2 interacts with the PI3 kinase Tor2, and in humans CLK-2 was recently shown to bind to and be required for the stability of all PI3 kinases, including ATR and ATM (Hayashi et al., 2007; Takai et al., 2007)
Mammalian p53 acts primarily as a transcription factor, and is essential for maintaining genome stability and triggering apoptosis in abnormal cells that might be predisposed to form tumors (Pierotti and Dragani, 1992; Roemer, 1999; Vogelstein et al., 2000). The central role of p53 as a tumor suppressor is demonstrated by the fact that most human cancers evolve ways to evade p53 tumor suppressor activity by selecting for mutations in p53 itself, inactivating upstream signaling components (Bartek and Lukas, 2003; Sharpless and DePinho, 1999), or over-expressing negative regulators such as Mdm2 (Freedman et al., 1999). The p53 homologs, termed p63 and p73, also impinge on DNA damage response regulation (Suh et al., 2006). While p53 does not have an obvious developmental role in mammals, p63 and p73 are strongly implicated in developmental control (for review, see (Muller et al., 2006).
The only known gene product that uniquely affects IR-induced apoptosis in C. elegans is CEP-1 (C. elegans p53-like), a primordial p53 family member (Table 2; Derry et al., 2001; Schumacher et al., 2001). Due to low sequence identity between CEP-1 and its homologs, which is mostly restricted to the DNA-binding domain, it has been unclear whether CEP-1 is most closely related to mammalian p53, p63, or p73. Given its role in DNA damage-induced apoptosis, CEP-1 was initially thought to be most closely related to p53, although its sequence is more similar to p63 (Suh et al., 2006). A relationship to p63 is corroborated by data suggesting that an ancestral p63 isoform controls DNA damage-induced apoptosis in the mouse germline (Suh et al., 2006). CEP-1 is able to bind, albeit with lower affinity, to human p53 consensus sites, and structural analysis indicates that some key features of its DNA binding are conserved (Huyen et al., 2004; Schumacher et al., 2001). Unlike mammalian p53, but similar to Drosophila p53, cep-1 is not required for IR-induced cell cycle arrest (Derry et al., 2001; Schumacher et al., 2001). However, more recent studies implicated cep-1 in UV-induced cell cycle arrest of mitotic germ cells, perhaps by transcriptionally inducing the phg-1 gene (Derry et al., 2007; Stergiou et al., 2007). The dual function for CEP-1 in IR and UV-induced apoptosis and UV-induced cell cycle arrest is consistent with the accumulation of CEP-1 protein in mitotic germ cells and late pachytene stage cells (Schumacher et al., 2005). In response to IR, CEP-1 induces transcription of genes encoding two BH3 domain only proteins, EGL-1 and CED-13 (Schumacher et al., 2005). Similarly, mammalian p53 transcriptionally induces BH3 only domain proteins, PUMA and Noxa (Oda et al., 2000; Yu et al., 2001). An attractive idea is that transcriptional induction of BH3 domain-only proteins by CEP-1/p53 is the central and conserved regulatory node through which DNA damage induces apoptosis.
Several negative regulators of cep-1 have been identified. Based on bioinformatics, it seems unlikely that the worm genome encodes a homolog of MDM-2, an E3 ligase and the most common negative regulator of p53 in mammals. However, the p53-binding protein IASP-1 (Bergamaschi et al., 2003) appears to be conserved. Both human IASP-1 and its worm homolog APE-1 are able to bind directly to p53 and CEP-1, respectively, and depleting iasp-1/ape-1 enhances p53 and cep-1-dependent apoptosis in the respective systems. Interestingly, however, while ape-1 inactivation leads to elevated cep-1-dependent apoptosis in the absence of genotoxic agents, increased apoptosis in response to DNA-damaging agents is not further increased by ape-1 RNAi, in apparent contrast to what might be expected from a negative regulator of cep-1 (see below).
Another gene that has been implicated in negatively regulating cep-1 is the abl-1 tyrosine kinase (Deng et al., 2004). Although no direct interaction between ABL-1 and either CEP-1 or any other upstream DNA damage checkpoint pathway component has been established, abl-1 acts specifically on DNA damage induced apoptosis. Interestingly, the phenotype of abl-1 knockouts can be mimicked by treating worms with chemical inhibitors of the ABL-1 kinase, including Gleevec(r) (imatinib mesylate), a drug used in human cancer therapy. Recent studies also confirmed a role of C. elegans akt-1 and akt-2 kinases, which had previously been implicated in insulin signaling, in down-regulating cep-1 (Quevedo et al., 2007). While disruption of both proteins leads to sterility, single knockouts elevate levels of cep-1-dependent apoptosis. These kinases seem to act upstream of cep-1, as their removal appears to result in elevation in CEP-1 abundance, posttranscriptional modification, or both.
The GLD-1 translational repressor, which controls several aspects of germline development, negatively regulates cep-1 expression (Schumacher et al., 2005). An unusual hypomorphic gld-1 mutation, op236, was isolated in an unbiased genetic screen for genes that decrease CEP-1 activity. GLD-1 binds directly to the cep-1 3'UTR and represses CEP-1 translation. Interestingly, whereas the GLD-1 op236 mutant protein binds poorly to the cep-1 3'UTR, it binds and regulates many developmental GLD-1 target mRNAs, which is consistent with the virtually wild type appearance of gld-1(op236) gonads. GLD-1 binding to the cep-1 3'-UTR is necessary and sufficient for cep-1 translational repression, as replacing the cep-1 3'UTR with an unrelated UTR renders cep-1 insensitive to GLD-1-mediated repression. It will be interesting to explore the role and function of other op236-like mutations in the future and to assess whether the human homolog of GLD-1, called quaking, similarly controls p53 expression.
In addition to classic genetic approaches that led to the discovery of the above described genes, functional genomics and RNAi based studies implicated several additional loci in DNA damage induced apoptosis (Boulton et al., 2002; van Haaften et al., 2006; Table 2). Furthermore, transcriptional coactivators previously thought to be dedicated to the Notch signaling pathway, called Mastermind-like (MAML)1-3 in mammals and LAG-3/SEL-8 in C. elegans, turned out to function as transcriptional coactivators of p53 and CEP-1, respectively (Zhao et al., 2007). However, an effect of these coactivators on the DNA damage response has not yet been tested by mutational analysis.
An important question is whether germ cell DNA damage checkpoints, cell cycle arrest, and apoptosis are critical for preventing mutagenesis or maintaining genome integrity under normal circumstances. To address this question, mutation rates in various checkpoint mutants have been assessed (Harris et al., 2006). In these experiments, the accumulation of mutations, which mostly comprised of small deletions, correlated with the degree of radiation sensitivity associated with the particular mutant tested, but not with its propensity for either DNA damage-induced apoptosis or cell cycle arrest. For example, mrt-1 and hus-1 mutants are highly sensitive to radiation, and showed the highest spontaneous mutation rate. In contrast, clk-2 alleles are less sensitive to ionizing irradiation and accumulated few or no spontaneous mutations. All three mutants are equally defective in DNA damage-induced cell cycle arrest and apoptosis. In addition, no mutator phenotype was detected in cep-1 mutants or in mutants defective in the core cell apoptosis pathway (Harris et al., 2006). Thus, checkpoint-mediated DNA repair seems to be by far the most important function of DNA damage checkpoint genes with respect to maintaining genome stability. Interestingly, however, checkpoint-induced apoptosis might be very important for monitoring meiotic recombination and chromosome pairing (see Section 7).
Besides DNA damage, defects in meiotic prophase checkpoints can also trigger apoptosis during pachytene. During meiotic prophase, homologous chromosomes pair and engage in meiotic recombination. These processes are monitored by distinct recombination and pairing checkpoints. The meiotic recombination checkpoint was uncovered by inactivation of key genes involved in double strand break repair and meiotic recombination, such as rad-51 and brca-2 (Alpi et al., 2003; Gartner et al., 2000; Martin et al., 2005). Inactivation of these genes results in germ cell apoptosis that requires the cep-1, clk-2 and mrt-1 checkpoint genes. Consistent with the recombination checkpoint being involved in monitoring meiotic recombination, the excessive levels of apoptosis occurring in rad-51 mutants are bypassed by inactivating the SPO-11 nuclease, which is required for generating meiotic DNA double strand breaks (Gartner et al., 2000). At present, pathways leading to double strand break-induced apoptosis and recombination failure-induced apoptosis cannot be genetically separated. This suggests that IR-induced germ cell apoptosis may derive primarily from double strand breaks.
Meiotic chromosome pairing occurs in several steps. Initial chromosome recognition and pairing, referred to as synapsis-independent pairing, is followed by synapsis, which is the intimate pairing and assembly of homologous chromosomes into a proteinacious scaffold termed the synaptonemal complex (SC). In most organisms, but not in D. melanogaster and C. elegans, pairing is dependent on meiotic double strand breaks generated by the SPO-11 nuclease (Dernburg et al., 1998). Because in C. elegans spo-11-dependent double strand breaks are not required for pairing, the recombination and pairing checkpoints are separate, each resulting in germ cell apoptosis (Bhalla and Dernburg, 2005), for review see (Meier and Gartner, 2006). Cis-acting mutations in the X-chromosome pairing center (PC), a region needed for efficient pairing initiation, trigger the DNA damage checkpoint and spo-11-independent apoptosis (Bhalla and Dernburg, 2005). Apoptosis was, however, blocked in pch-2 mutants, which do not affect DNA damage-induced apoptosis (Bhalla and Dernburg, 2005). Thus, the failure to pair might be sensed by a pch-2 dependent pairing checkpoint, and that checkpoint may in turn lower the frequency of meiotic nondisjunction. Indeed, the nondisjunction frequency of mutants hemizygous for the X chromosome PC is enhanced in a ced-4 apoptosis defective background (Bhalla and Dernburg, 2005).
The DNA damage checkpoint is activated in him-8 mutants that are defective in X-chromosome pairing and in mutants containing a PC deletion on chromosomes. This may be explained by accumulation of unprocessed meiotic double strand breaks that are generated by SPO-11, but cannot be repaired by gene conversion due to a failure in pairing (MacQueen et al., 2005). In line with the notion that the pachytene checkpoint may deal with meiotic double strand breaks, the frequency with which chromosomes fail to pair in him-8 hermaphrodites is enhanced in ced-4 apoptosis-defective mutants (MacQueen et al., 2005). Thus, defects in both meiotic recombination and pairing might lead to elimination of compromised cells, which under natural conditions might be the most important function of checkpoint-dependent pachytene cell apoptosis. In this context, it is interesting to note that CEP-1 abundance is inversely correlated with ongoing recombination: CEP-1 is absent in transition zone nuclei where recombination is initiated and is actively taking place, and high in late pachytene nuclei that have completed recombination and where apoptosis occurs. (Alpi et al., 2003; Schumacher et al., 2005). Thus, CEP-1 abundance might be regulated so that apoptosis cannot be triggered by double strand breaks that occur naturally during meiotic recombination, but can be induced in late pachytene cells if recombination intermediates are not repaired in a timely manner.
In addition to the DNA integrity checkpoint described above, germ cell apoptosis is also increased by exposure to certain environmental stresses and particular pathogens (Aballay and Ausubel, 2001; Salinas et al., 2006). As is true for DNA damage, these stresses do not induce apoptosis of somatic cells, but instead specifically increase apoptosis during oogenesis.
Various environmental stresses that do not directly compromise genomic integrity (oxidative, heat, salt, starvation) nevertheless induce germ cell apoptosis (Salinas et al., 2006). Apoptosis induced by these stresses does not require either egl-1 or cep-1, indicating that the physiological apoptosis pathway or a parallel egl-1-independent mechanism is involved (Figure 2). The induction of apoptosis by oxidative, heat, or osmotic stress depends upon the redundant activities of the conserved p38 and JNK stress-activated protein kinase pathways, as indicated by overlapping requirements for the corresponding MAPK kinases, SEK-1 and MEK-1, respectively. This cell death induction also requires abl-1, an intriguing finding given that abl-1 antagonizes cep-1-mediated induction of germ cell apoptosis (see Section 6). Starvation induces germ cell apoptosis independently of MEK-1, SEK-1 and ABL-1, and therefore must act through a different mechanism. Because these environmental stresses do not affect genomic integrity, each is likely to place severe demands on the oocyte cytoplasm. If the dying oocyte nuclei act as “nurse cells” under these stress conditions (Section 4.1), the increased apoptosis might buy time, during which damaged components can be repaired or re-synthesized before incorporation into oocytes. Further analysis of how these “cytoplasmic” stresses influence developmental germ cell apoptosis may yield important insights not only into how germ cells respond to adversity, but also into how physiological germ cell apoptosis is normally regulated.
Germ cell apoptosis is also readily activated in response to infections with pathogenic Salmonella typhimurium strains (Aballay and Ausubel, 2001). This pathogenesis-induced germ cell apoptosis depends on signaling through SEK-1 and its downstream p38 kinase PMK-1 (Aballay et al., 2003). Interestingly, this apoptosis also requires egl-1 (Aballay and Ausubel, 2001), indicating that p38 signaling can promote germ cell apoptosis through egl-1-dependent (pathogenesis) and -independent (environmental stress) pathways. The role of p38 signaling in germ cell apoptosis thus appears to be highly complex, a notion that is reinforced by the finding that the p38 kinase PMK-3 is required to prevent excessive physiological apoptosis (Lettre et al., 2004). For the stresses discussed so far in this chapter, it is thought that increases in apoptosis ultimately preserve the functionality and integrity of oogenesis. Interestingly, if the core apoptosis machinery is disabled, C. elegans are less likely to survive Salmonella typhimurium infection, suggesting that under these conditions germ cell apoptosis enhances the fitness of the whole organism (Aballay and Ausubel, 2001). Perhaps this apoptosis allows key metabolic resources to be diverted towards defending against the pathogen attack. Germ cell ablation increases C. elegans lifespan under non-stressed conditions, by acting through the somatic gonad and intestine in an endocrine pathway (Berman and Kenyon, 2006). During pathogen infection, germ cell apoptosis might promote survival through a similar systemic mechanism.
Germ cells and somatic cells are likely to be subject to distinct selective pressures, as somatic cells are optimized to contribute to the fitness for one generation, whereas germ cells are optimized for both the maintenance of eternal proliferative potential and the accurate transmission of the genetic material. This dichotomy is most extreme in the worm life cycle, as adult somatic cells are entirely postmitotic and invariantly derive from the feritilized zygote by a limited number of cell cycles, rather than being continuously replenished by adult somatic stem cells as in most other animals. Thus, while somatic tissues may tolerate high levels of DNA damage, germline-specific checkpoints might have evolved to guard germ cells from acquiring deleterious mutations that could be passed on to the next generation.
Recent studies have addressed the connection between DNA damage checkpoint pathways, germ cell apoptosis and DNA repair factors, and the maintenance of immortal germlines (Ahmed, 2006). Unbiased genetic screens have identified mutants that are defective in germ cell immortality (Ahmed and Hodgkin, 2000). These mrt (mortal germline) mutants proliferate normally for several generations, before eventually becoming sterile due to defects in germ cell proliferation. Several of these mutants have been implicated in telomere length maintenance (Ahmed and Hodgkin, 2000; Meier et al., 2006). Their mortal germ cell phenotype can be explained by a loss of telomerase activity. This defect results in progressive telomere attrition, which leads in turn (after 10 to 15 worm generations) to critically short telomeres that may engage in chromosomal fusion events and concomitant genome instability. Besides telomerase, which is not required for DNA damage responses, genes encoding the 9-1-1 DNA damage checkpoint complex (e.g. mrt-2 and hus-1) are needed for telomere length maintenance (Ahmed and Hodgkin, 2000; Boerckel et al., 2007; Hofmann et al., 2002; Meier et al., 2006). The mammalian hus-1 ortholog was recently also implicated in telomere replication, and is likely to be required for telomerase loading (Francia et al., 2006). In addition to 9-1-1 complex components, the C. elegans him-6 gene, which is a homolog of the Blooms syndrome gene and apparently not involved in controlling telomere length, was also implicated in germ cell mortality (Ahmed, 2006; Grabowski et al., 2005). In this last case, excess apoptosis presumably occurs due to accumulation of excessive DNA damage.
The above observations suggest that germ cell apoptosis may contribute to immortality by eliminating compromised germ cells. However, germ cell immortality does not require the elimination of faulty cells by apoptosis, at least under standard laboratory conditions, as cep-1, ced-3, and ced-4 mutations are not mortal (Ahmed, 2006). Similarly, no obvious correlation exists between postmitotic aging phenotypes and germ cell mortality, as short lived daf-16 and daf-18 mutants do not have a mortal germline phenotype and as telomerase mutations in turn do not affect postmitotic longevity (Ahmed, 2006). Given that many mortal germ cell mutations do not seem to affect telomere length and DNA repair, it will be interesting to identify additional mechanisms needed for maintaining immortal germ cells.
Apoptosis plays a prominent role during normal C. elegans oogenesis, and provides a means for eliminating oocytes with defective genomes. Thus, germ cell apoptosis is important not only for optimizing production and allocation of precious oocyte cytoplasmic contents, but also for maintaining genomic integrity for the next generation. The observation that developing C. elegans oocytes respond to environmental and genotoxic stress by undergoing apoptosis illustrates the uniqueness of the germline with respect to its need to eliminate genetically compromised cells.
Further analyses of germ cell apoptosis in C. elegans are likely to yield a number of important and interesting new findings. We can now be reasonably confident that physiological germ cell apoptosis optimizes the oogenesis process, but it remains of intense interest to elucidate how this cell death is regulated. Physiological germ cell apoptosis is induced by an important control point during oocyte development, and a novel and as yet unknown mechanism for activating the core apoptosis machinery. In addition, a mechanistic understanding of how cep-1-independent germ cell apoptosis is increased by environmental stress, and by apoptosis genes such as those listed in Table 1, will identify processes and regulatory signals that are important for oogenesis. These mechanisms are likely to be relevant to the apoptosis that occurs in normal developing oocytes in mammals. With respect to DNA damage-induced apoptosis, now that the basic framework for the genome integrity checkpoint has been uncovered, it will be exciting to employ the genetic advantages of C. elegans to elucidate novel processes and mechanisms that are involved in this checkpoint. These mechanisms are also highly likely to be conserved. Finally, C. elegans will provide a powerful genetic system for identifying and probing mechanisms that are important for maintaining immortality of the germline, a biological process of fundamental importance. Much has been learned since the first description of C. elegans germ cell apoptosis was published in 1999. The fascinating complexity of what we now know about these apoptosis pathways suggests that this field will keep scientists busy for many years to come.
Preparation of this chapter was supported by the Harvard Stem Cell Institute (TKB), the Myra R. Reinhard Family Foundation (PRB), a Cancer Research UK career development award fellowship (AG) and an AICR project grant (AG). We thank Rachael Rutkowski for helpful comments and Melissa Pepling and Frank McKeon for helpful discussions about oocyte apoptosis in the mouse.
Aballay, A., and Ausubel, F.M. (2001). Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc. Natl. Acad. Sci. U.S.A. 98, 2735–2739. Abstract Article
Aballay, A., Drenkard, E., Hilbun, L.R., and Ausubel, F.M. (2003). Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 13, 47–52. Abstract Article
Ahmed, S. (2006). Uncoupling of pathways that promote postmitotic life span and apoptosis from replicative immortality of Caenorhabditis elegans germ cells. Aging Cell 5, 559–563. Abstract Article
Ahmed, S., Alpi, A., Hengartner, M.O., and Gartner, A. (2001). C. elegans RAD-5/CLK-2 defines a new DNA damage checkpoint protein. Curr. Biol. 11, 1934–1944. Abstract Article
Ahmed, S., and Hodgkin, J. (2000). MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature 403, 159–164. Abstract Article
Alpi, A., Pasierbek, P., Gartner, A., and Loidl, J. (2003). Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma 112, 6–16. Abstract Article
Audhya, A., Hyndman, F., McLeod, I.X., Maddox, A.S., Yates, J.R., 3rd, Desai, A., and Oegema, K. (2005). A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J. Cell Biol. 171, 267–279. Abstract Article
Baker, T.G. (1963). A quantitative and cytological study of germ cells and human ovaries. Proc. R. Soc. Lond. 158, 417–433. Abstract
Barbee, S.A., Estes, P.S., Cziko, A.M., Hillebrand, J., Luedeman, R.A., Coller, J.M., Johnson, N., Howlett, I.C., Geng, C., Ueda, R., et al. (2006). Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron 52, 997–1009. Abstract Article
Bartek, J., and Lukas, J. (2003). Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429. Abstract Article
Benard, C., McCright, B., Zhang, Y., Felkai, S., Lakowski, B., and Hekimi, S. (2001). The C. elegans maternal-effect gene clk-2 is essential for embryonic development, encodes a protein homologous to yeast Tel2p and affects telomere length. Development 128, 4045–4055. Abstract
Bergamaschi, D., Samuels, Y., O'Neil, N.J., Trigiante, G., Crook, T., Hsieh, J.K., O'Connor, D.J., Zhong, S., Campargue, I., Tomlinson, M.L., et al. (2003). iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat. Genet. 33, 162–167. Abstract Article
Berman, J.R., and Kenyon, C. (2006). Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124, 1055–1068. Abstract Article
Bhalla, N., and Dernburg, A.F. (2005). A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science 310, 1683–1686. Abstract Article
Boag, P.R., Nakamura, A., and Blackwell, T.K. (2005). A conserved RNA-protein complex component involved in physiological germline apoptosis regulation in C. elegans. Development 132, 4975–4986. Abstract Article
Boerckel, J., Walker, D., and Ahmed, S. (2007). The C. elegans Rad17 homolog HPR-17 is required for telomere replication. Genetics 176, 703–709. Abstract Article
Borum, K. (1961). Oogenesis in the mouse. A study of the meiotic prophase. Exp. Cell Res. 24, 495–507. Abstract Article
Boulton, S.J., Gartner, A., Reboul, J., Vaglio, P., Dyson, N., Hill, D.E., and Vidal, M. (2002). Combined functional genomic maps of the C. elegans DNA damage response. Science 295, 127–131. Abstract Article
Boulton, S.J., Martin, J.S., Polanowska, J., Hill, D.E., Gartner, A., and Vidal, M. (2004). BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr. Biol. 14, 33–39. Abstract Article
Chin, G.M., and Villeneuve, A.M. (2001). C. elegans mre-11 is required for meiotic recombination and DNA repair but is dispensable for the meiotic G2 DNA damage checkpoint. Genes Dev. 15, 522–534. Abstract Article
Cho, S., Rogers, K.W., and Fay, D.S. (2007). The C. elegans glycopeptide hormone receptor ortholog, FSHR-1, regulates germline differentiation and survival. Curr. Biol. 17, 203–212. Abstract Article
Church, D.L., Guan, K.L., and Lambie, E.J. (1995). Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 121, 2525–2535. Abstract
Collis, S.J., Barber, L.J., Clark, A.J., Martin, J.S., Ward, J.D., and Boulton, S.J. (2007). HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat. Cell Biol. 9, 391–401. Abstract Article
Costa, R.M., Chigancas, V., Galhardo Rda, S., Carvalho, H., and Menck, C.F. (2003). The eukaryotic nucleotide excision repair pathway. Biochimie 85, 1083–1099. Abstract Article
Decker, C.J., and Parker, R. (2006). CAR-1 and trailer hitch: driving mRNP granule function at the ER? J. Cell Biol. 173, 159–163. Abstract Article
Deng, X., Hofmann, E.R., Villanueva, A., Hobert, O., Capodieci, P., Veach, D.R., Yin, X., Campodonico, L., Glekas, A., Cordon-Cardo, C., et al. (2004). Caenorhabditis elegans ABL-1 antagonizes p53-mediated germline apoptosis after ionizing irradiation. Nat. Genet. 36, 906–912. Abstract Article
Dengg, M., Garcia-Muse, T., Gill, S.G., Ashcroft, N., Boulton, S.J., and Nilsen, H. (2006). Abrogation of the CLK-2 checkpoint leads to tolerance to base-excision repair intermediates. EMBO Rep. 7, 1046–1051. Abstract Article
Dernburg, A.F., McDonald, K., Moulder, G., Barstead, R., Dresser, M., and Villeneuve, A.M. (1998). Meiotic recombination in C. elegans initiates by a conserved mechanism and is dispensable for homologous chromosome synapsis. Cell 94, 387–398. Abstract Article
Derry, W.B., Bierings, R., van Iersel, M., Satkunendran, T., Reinke, V., and Rothman, J.H. (2007). Regulation of developmental rate and germ cell proliferation in Caenorhabditis elegans by the p53 gene network. Cell Death Differ. 14, 662–670. Abstract Article
Derry, W.B., Putzke, A.P., and Rothman, J.H. (2001). Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294, 591–595. Abstract Article
Francia, S., Weiss, R.S., Hande, M.P., Freire, R., and d'Adda di Fagagna, F. (2006). Telomere and telomerase modulation by the mammalian Rad9/Rad1/Hus1 DNA-damage-checkpoint complex. Curr. Biol. 16, 1551–1558. Abstract Article
Freedman, D.A., Wu, L., and Levine, A.J. (1999). Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 55, 96–107. Abstract Article
Garcia-Muse, T., and Boulton, S.J. (2005). Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J. 24, 4345–4355. Abstract Article
Gartner, A., MacQueen, A.J., and Villeneuve, A.M. (2004). Methods for analyzing checkpoint responses in Caenorhabditis elegans. Methods Mol. Biol. 280, 257–274. Abstract
Gartner, A., Milstein, S., Ahmed, S., Hodgkin, J., and Hengartner, M.O. (2000). A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5, 435–443. Abstract Article
Grabowski, M.M., Svrzikapa, N., and Tissenbaum, H.A. (2005). Bloom syndrome ortholog HIM-6 maintains genomic stability in C. elegans. Mech. Ageing Dev. 126, 1314–1321. Abstract Article
Gumienny, T.L., Lambie, E., Hartwieg, E., Horvitz, H.R., and Hengartner, M.O. (1999). Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126, 1011–1022. Abstract
Hajnal, A., and Berset, T. (2002). The C. elegans MAPK phosphatase LIP-1 is required for the G2/M meiotic arrest of developing oocytes. EMBO J. 21, 4317–4326. Abstract Article
Harris, J., Lowden, M., Clejan, I., Tzoneva, M., Thomas, J.H., Hodgkin, J., and Ahmed, S. (2006). Mutator phenotype of Caenorhabditis elegans DNA damage checkpoint mutants. Genetics 174, 601–616. Abstract Article
Hartman, P.S., and Herman, R.K. (1982). Radiation-sensitive mutants of Caenorhabditis elegans. Genetics 102, 159–178. Abstract
Hasegawa, E., Karashima, T., Sumiyoshi, E., and Yamamoto, M. (2006). C. elegans CPB-3 interacts with DAZ-1 and functions in multiple steps of germline development. Dev. Biol. 295, 689–699. Abstract Article
Hayashi, T., Hatanaka, M., Nagao, K., Nakaseko, Y., Kanoh, J., Kokubu, A., Ebe, M., and Yanagida, M. (2007). Rapamycin sensitivity of the Schizosaccharomyces pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells 12, 1357–1370. Abstract Article
Hofmann, E.R., Milstein, S., Boulton, S.J., Ye, M., Hofmann, J.J., Stergiou, L., Gartner, A., Vidal, M., and Hengartner, M.O. (2002). Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr. Biol. 12, 1908–1918. Abstract Article
Holway, A.H., Kim, S.H., La Volpe, A., and Michael, W.M. (2006). Checkpoint silencing during the DNA damage response in Caenorhabditis elegans embryos. J. Cell Biol. 172, 999–1008. Abstract Article
Huyen, Y., Jeffrey, P.D., Derry, W.B., Rothman, J.H., Pavletich, N.P., Stavridi, E.S., and Halazonetis, T.D. (2004). Structural differences in the DNA binding domains of human p53 and its C. elegans ortholog CEP-1. Structure 12, 1237–1243. Abstract Article
Kalogeropoulos, N., Christoforou, C., Green, A.J., Gill, S., and Ashcroft, N.R. (2004). chk-1 is an essential gene and is required for an S-M checkpoint during early embryogenesis. Cell Cycle 3, 1196–1200. Abstract
Karashima, T., Sugimoto, A., and Yamamoto, M. (2000). Caenorhabditis elegans homologue of the human azoospermia factor DAZ is required for oogenesis but not for spermatogenesis. Development 127, 1069–1079. Abstract
Kinchen, J.M., Doukoumetzidis, K., Almendinger, J., Stergiou, L., Tosello-Trampont, A., Sifri, C.D., Hengartner, M.O., and Ravichandran, K.S. (2008). A pathway for phagosome maturation during engulfment of apoptotic cells. Nat. Cell Biol. 10, 556–566. Abstract Article
Kota, R.S., and Runge, K.W. (1999). Tel2p, a regulator of yeast telomeric length in vivo, binds to single-stranded telomeric DNA in vitro. Chromosoma 108, 278–290. Abstract Article
Krakauer, D.C., and Mira, A. (1999). Mitochondria and germ-cell death. Nature 400, 125–126. Abstract Article
Kritikou, E.A., Milstein, S., Vidalain, P.O., Lettre, G., Bogan, E., Doukoumetzidis, K., Gray, P., Chappell, T.G., Vidal, M., and Hengartner, M.O. (2006). C. elegans GLA-3 is a novel component of the MAP kinase MPK-1 signaling pathway required for germ cell survival. Genes Dev. 20, 2279–2292. Abstract Article
Lackner, M.R., Kindt, R.M., Carroll, P.M., Brown, K., Cancilla, M.R., Chen, C., de Silva, H., Franke, Y., Guan, B., Heuer, T., et al. (2005). Chemical genetics identifies Rab geranylgeranyl transferase as an apoptotic target of farnesyl transferase inhibitors. Cancer Cell 7, 325–336. Abstract Article
Leacock, S.W., and Reinke, V. (2006). Expression profiling of MAP kinase-mediated meiotic progression in Caenorhabditis elegans. PLoS Genet. 2, e174. Abstract Article
Lee, M.H., Hook, B., Lamont, L.B., Wickens, M., and Kimble, J. (2006). LIP-1 phosphatase controls the extent of germline proliferation in Caenorhabditis elegans. EMBO J. 25, 88–96. Abstract Article
Lettre, G., and Hengartner, M.O. (2006). Developmental apoptosis in C. elegans: a complex CEDnario. Nat. Rev. Mol. Cell Biol. 7, 97–108. Abstract Article
Lettre, G., Kritikou, E.A., Jaeggi, M., Calixto, A., Fraser, A.G., Kamath, R.S., Ahringer, J., and Hengartner, M.O. (2004). Genome-wide RNAi identifies p53-dependent and -independent regulators of germ cell apoptosis in C. elegans. Cell Death Differ. 11, 1198–1203. Abstract Article
MacQueen, A.J., Phillips, C.M., Bhalla, N., Weiser, P., Villeneuve, A.M., and Dernburg, A.F. (2005). Chromosome sites play dual roles to establish homologous synapsis during meiosis in C. elegans. Cell 123, 1037–1050. Abstract Article
MacQueen, A.J., and Villeneuve, A.M. (2001). Nuclear reorganization and homologous chromosome pairing during meiotic prophase require C. elegans chk-2. Genes Dev. 15, 1674–1687. Abstract Article
Martin, J.S., Winkelmann, N., Petalcorin, M.I., McIlwraith, M.J., and Boulton, S.J. (2005). RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol. Cell. Biol. 25, 3127–3139. Abstract Article
Meier, B., Clejan, I., Liu, Y., Lowden, M., Gartner, A., Hodgkin, J., and Ahmed, S. (2006). trt-1 is the Caenorhabditis elegans catalytic subunit of telomerase. PLoS Genet. 2, e18. Abstract Article
Meier, B., and Gartner, A. (2006). Meiosis: checking chromosomes pair up properly. Curr. Biol. 16, R249–R251. Abstract Article
Muller, M., Schleithoff, E.S., Stremmel, W., Melino, G., Krammer, P.H., and Schilling, T. (2006). One, two, three-p53, p63, p73 and chemosensitivity. Drug Resist. Updat. 9, 288–306. Abstract Article
Navarro, R.E., Shim, E.Y., Kohara, Y., Singson, A., and Blackwell, T.K. (2001). cgh-1, a conserved predicted RNA helicase required for gametogenesis and protection from physiological germline apoptosis in C. elegans. Development 128, 3221–3232. Abstract
Oda, E., Ohki, R., Murasawa, H., Nemoto, J., Shibue, T., Yamashita, T., Tokino, T., Taniguchi, T., and Tanaka, N. (2000). Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058. Abstract Article
Park, D., Jia, H., Rajakumar, V., and Chamberlin, H.M. (2006). Pax2/5/8 proteins promote cell survival in C. elegans. Development 133, 4193–4202. Abstract Article
Pepling, M.E. (2006). From primordial germ cell to primordial follicle: mammalian female germ cell development. Genesis 44, 622–632. Abstract Article
Pepling, M.E., and Spradling, A.C. (2001). Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev. Biol. 234, 339–351. Abstract Article
Pierotti, M.A., and Dragani, T.A. (1992). Genetics and cancer. Curr. Opin. Oncol. 4, 127–133. Abstract Article
Prakash, S., and Prakash, L. (2000). Nucleotide excision repair in yeast. Mutat. Res. 451, 13–24. Abstract
Quevedo, C., Kaplan, D.R., and Derry, W.B. (2007). AKT-1 regulates DNA-damage-induced germline apoptosis in C. elegans. Curr. Biol. 17, 286–292. Abstract Article
Roemer, K. (1999). Mutant p53: gain-of-function oncoproteins and wild-type p53 inactivators. Biol. Chem. 380, 879–887. Abstract Article
Salinas, L.S., Maldonado, E., and Navarro, R.E. (2006). Stress-induced germ cell apoptosis by a p53 independent pathway in Caenorhabditis elegans. Cell Death Differ. 13, 2129–2139. Abstract Article
Schertel, C., and Conradt, B. (2007). C. elegans orthologs of components of the RB tumor suppressor complex have distinct pro-apoptotic functions. Development 134, 3691–3701. Abstract Article
Schumacher, B., Hanazawa, M., Lee, M.H., Nayak, S., Volkmann, K., Hofmann, E.R., Hengartner, M., Schedl, T., and Gartner, A. (2005). Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 120, 357–368. Abstract Article
Schumacher, B., Hofmann, K., Boulton, S., and Gartner, A. (2001). The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 11, 1722–1727. Abstract Article
Schumacher, B., Schertel, C., Wittenburg, N., Tuck, S., Mitani, S., Gartner, A., Conradt, B., and Shaham, S. (2005). C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ. 12, 153–161. Abstract Article
Schwartz, H.T. (2007). A protocol describing pharynx counts and a review of other assays of apoptotic cell death in the nematode worm Caenorhabditis elegans. Nat. Protoc. 2, 705–714. Abstract Article
Sharpless, N.E., and DePinho, R.A. (1999). The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 9, 22–30. Abstract Article
Shikata, M., Ishikawa, F., and Kanoh, J. (2007). Tel2 is required for activation of the Mrc1-mediated replication checkpoint. J. Biol. Chem. 282, 5346–5355. Abstract Article
Squirrell, J.M., Eggers, Z.T., Luedke, N., Saari, B., Grimson, A., Lyons, G.E., Anderson, P., and White, J.G. (2006). CAR-1, a protein that localizes with the mRNA decapping component DCAP-1, is required for cytokinesis and ER organization in Caenorhabditis elegans embryos. Mol. Biol. Cell 17, 336–344. Abstract Article
Stergiou, L., Doukoumetzidis, K., Sendoel, A., and Hengartner, M.O. (2007). The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differ. 14, 1129–1138. Abstract Article
Suh, E.K., Yang, A., Kettenbach, A., Bamberger, C., Michaelis, A.H., Zhu, Z., Elvin, J.A., Bronson, R.T., Crum, C.P., and McKeon, F. (2006). p63 protects the female germ line during meiotic arrest. Nature 444, 624–628. Abstract Article
Takai, H., Wang, R.C., Takai, K.K., Yang, H., and de Lange, T. (2007). Tel2 regulates the stability of PI3K-related protein kinases. Cell 131, 1248–1259. Abstract Article
van Haaften, G., Romeijn, R., Pothof, J., Koole, W., Mullenders, L.H., Pastink, A., Plasterk, R.H., and Tijsterman, M. (2006). Identification of conserved pathways of DNA-damage response and radiation protection by genome-wide RNAi. Curr. Biol. 16, 1344–1350. Abstract Article
Vogelstein, B., Lane, D., and Levine, A.J. (2000). Surfing the p53 network. Nature 408, 307–310. Abstract Article
Weston, A., and Sommerville, J. (2006). Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res. 34, 3082–3094. Abstract Article
Whetstine, J.R., Nottke, A., Lan, F., Huarte, M., Smolikov, S., Chen, Z., Spooner, E., Li, E., Zhang, G., Colaiacovo, M., and Shi, Y. (2006). Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125, 467–481. Abstract Article
Wilczynska, A., Aigueperse, C., Kress, M., Dautry, F., and Weil, D. (2005). The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J. Cell. Sci. 118, 981–992. Abstract Article
Wilhelm, J.E., Buszczak, M., and Sayles, S. (2005). Efficient protein trafficking requires trailer hitch, a component of a ribonucleoprotein complex localized to the ER in Drosophila. Dev. Cell 9, 675–685. Abstract Article
Wolke, U., Jezuit, E.A., and Priess, J.R. (2007). Actin-dependent cytoplasmic streaming in C. elegans oogenesis. Development 134, 2227–2236. Abstract Article
*Edited by Judith Kimble and Susan Strome. Last revised June 3, 2008. Published September 4, 2008. This chapter should be cited as: Gartner et al. Germline Survival and Apoptosis (September 4, 2008), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.145.1, http://www.wormbook.org.
Copyright: © 2008 Anton Gartner et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Tel 0044-(0)1382-385809. E-mail: a.gartner@dundee.ac.uk
†To whom correspondence should be addressed. Tel 001-617-264-2760. E-mail: keith.blackwell@joslin.harvard.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.