Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Heterotrimeric G proteins, composed of α, β, and γ subunits, are able to transduce signals from membrane receptors to a wide variety of intracellular effectors. In this role, G proteins effectively function as dimers since the signal is communicated either by the Gα subunit or the stable Gβγ complex. When inactive, Gα-GDP associates with Gβγ and the cytoplasmic portion of the receptor. Ligand activation of the receptor stimulates an exchange of GTP for GDP resulting in the active signaling molecules Gα-GTP and free Gβγ, either of which can interact with effectors. Hydrolysis of GTP restores Gα-GDP, which then reassociates with Gβγ and receptor to terminate signaling. The rate of G protein activation can be enhanced by the guanine-nucleotide exchange factor, RIC-8, while the rate of GTP hydrolysis can be enhanced by RGS proteins such as EGL-10 and EAT-16. Evidence for a receptor-independent G-protein-signaling pathway has been demonstrated in C. elegans early embryogenesis. In this pathway, the Gα subunits GOA-1 and GPA-16 are apparently activated by the non-transmembrane proteins GPR-1, GPR-2, and RIC-8, and negatively regulated by RGS-7. The C. elegans genome encodes 21 Gα, 2 Gβ and 2 Gγ subunits. The α subunits include one ortholog of each mammalian Gα family: GSA-1 (Gs), GOA-1 (Gi/o), EGL-30 (Gq) and GPA-12 (G12). The remaining C. elegans α subunits (GPA-1, GPA-2, GPA-3, GPA-4, GPA-5, GPA-6, GPA-7, GPA-8, GPA-9, GPA-10, GPA-11, GPA-13, GPA-14, GPA-15, GPA-16, GPA-17 and ODR-3) are most similar to the Gi/o family, but do not share sufficient homology to allow classification. The conserved Gα subunits, with the exception of GPA-12, are expressed broadly while 14 of the new Gα genes are expressed in subsets of chemosensory neurons. Consistent with their expression patterns, the conserved C. elegans α subunits, GSA-1, GOA-1 and EGL-30 are involved in diverse and fundamental aspects of development and behavior. GOA-1 acts redundantly with GPA-16 in positioning of the mitotic spindle in early embryos. EGL-30 and GSA-1 are required for viability starting from the first larval stage. In addition to their roles in development and behaviors such as egg laying and locomotion, the EGL-30, GSA-1 and GOA-1 pathways interact in a network to regulate acetylcholine release by the ventral cord motor neurons. EGL-30 provides the core signals for vesicle release, GOA-1 negatively regulates the EGL-30 pathway, and GSA-1 modulates this pathway, perhaps by providing positional cues. Constitutively activated GPA-12 affects pharyngeal pumping. The Gα subunits unique to C. elegans are primarily involved in chemosensation. The Gβ subunit, GPB-1, as well as the Gγ subunit, GPC-2, appear to function along with the α subunits in the classic G protein heterotrimer. The remaining Gβ subunit, GPB-2, is thought to regulate the function of certain RGS proteins, while the remaining Gγ subunit, GPC-1, has a restricted role in chemosensation. The functional difference for most G protein pathways in C. elegans, therefore, resides in the α subunit. Many cells in C. elegans express multiple Gα subunits, and multiple G protein pathways are known to function in specific cell types. For example, Go, Gq and Gs-mediated signaling occurs in the ventral cord motor neurons. Similarly, certain amphid neurons use multiple G protein pathways to both positively and negatively regulate chemosensation. C. elegans thus provides a powerful model for the study of interactions between and regulation of G protein signaling.
Heterotrimeric G proteins, composed of α, β, and γ subunits, transduce signals from the plasma membrane through a cycle of guanine nucleotide exchange and hydrolysis. When signaling, G proteins effectively function as dimers since the signal is communicated either by the Gα subunit or the stable Gβγ complex. The classical G protein cycle is shown in Figure 1. Gα-GDP associates with Gβγ and with cytoplasmic portions of its typically seven-pass membrane receptor (also known as G protein coupled receptors; GPCRs). Upon ligand activation, the receptor acts as a guanine-nucleotide exchange factor (GEF) stimulating the exchange of GDP for GTP on the α subunit. Gα-GTP dissociates from Gβγ and both entities can interact with effectors. Hydrolysis of GTP restores Gα-GDP, which then reassociates with Gβγ and receptor to terminate signaling. In some pathways, Gα-GDP may interact with an additional GEF protein, RIC-8, which may then re-activate the α subunit (Tall et al., 2003; Reynolds et al., 2005). The rate of GTP hydrolysis can be enhanced by RGS proteins (regulator of G protein signaling; Watson et al. 1996). GEF proteins therefore act as positive regulators of G protein pathways while RGS proteins act as negative regulators (Figure 1).
G protein signaling can also be activated through a receptor-independent pathway in which the dissociation of Gα and Gβγ may be facilitated by GPR (G protein regulator) proteins, which compete with Gβγ for binding to the GDP-bound form of Gα and function as guanine nucleotide dissociation inhibitors (GDIs; Manning, 2003). Dissociation of Gα and Gβγ may also be facilitated by GEF proteins (Afshar at al., 2004). Alternatively, GEF proteins may recognize the Gα-GPR complex, and subsequently effect nucleotide exchange to generate an active Gα-GTP signaling molecule (Manning, 2003; Hess et al., 2004). Details of this pathway are discussed further under “Receptor-independent pathway” in the Gαo section.
|
Figure 1. Depicted is the activation of a heterotrimeric G protein by a G protein coupled receptor (GPCR). In the inactive conformation (1), the Gαβγ heterotrimer is associated with a seven transmembrane-spanning receptor protein, and the Gα subunit is associated with GDP. Upon receptor activation with ligand (*), the receptor undergoes a conformational change and acts as a guanine nucleotide exchange factor (GEF) for the α subunit, stimulating the exchange of GDP for GTP (2). Additional GEFs have been isolated that act on GDP associated Gα subunits not associated with receptor. Upon binding of GTP, the Gα subunit dissociates from the Gβγ dimmer and these proteins are released from the receptor. Dissociated Gα and Gβγ subunits then interact with a variety of effector (E) proteins (3). The signal is attenuated by the intrinsic ability of Gα subunits to hydrolyze GTP, and this activity is accelerated by GTPase-activating proteins (GAPs) (4). Most GAPs contain a regulator of G protein signaling (RGS) domain that often displays higher affinity for the Gα-GTP transition state.
C. elegans has 21 Gα, 2 Gβ and 2 Gγ genes (Jansen et al., 1999; Cuppen et al., 2003).
Based on sequence similarity, mammalian Gα subunits have been divided into four families: Gs, Gi/o, Gq and G12 (Neves et al., 2002). C. elegans expresses one ortholog of each of the mammalian families: GSA-1 (Gs), GOA-1 (Gi/o), EGL-30 (Gq) and GPA-12 (G12). The remaining C. elegans α subunits (GPA-1-11, GPA-13-17 and ODR-3) do not share sufficient homology to allow classification. The conserved Gα subunits, with the exception of GPA-12, are expressed broadly while 14 of the new Gα genes are expressed in subsets of chemosensory neurons (See Table 1).
Table 1. Expression patterns for G protein subunits.
| Gene | Amphid neurons | Other sensory neurons | Other neurons | Muscles | Pharynx | Other cells/ tissues | Embryonic | Subcellular | Reference |
|---|---|---|---|---|---|---|---|---|---|
| gsa-1 | all | All, male specific | All | Body wall, pharyngeal, vulval, male specific | Muscles, neurons | Excretory cell, intestine | Extensive | Korswagen et al., 1997; Park et al., 1997 | |
| egl-30 | ? | ? | Most | Pharyngeal, vulval, anal sphincter | Muscles | Sperm | Early embryos | Cell periphery in early embryos, neural cell bodies and processes, highest in axons of nerve ring | Lackner et al., 1999; Bastiani et al., 2003 |
| goa-1 | All | All | All | Pharyngeal, intestinal, vulval, uterine, male diagonal | Muscles, neurons | Spermatheca, distal tip cells | Early embryos | Cell membranes and centrosomes in embryo, neuronal processes in adults | Mendel et al., 1995; Ségalat et al., 1995; Miller and Rand, 2000 |
| gpa-1 | ADL, ASH, ASI, ASJ | PHA, PHB, SPD, SPV, SPC | Jansen et al., 1999; Jiang and Sternberg, 1999 | ||||||
| gpa-2 | AWC | PHA, PHB, and IL1L, IL2L, OLL or URB | PVT, AIA | Anal sphincter | M1, M5, I5 | Cilia, cell bodies, axons | Zwaal et al., 1997; Lans et al., 2004 | ||
| gpa-3 | ADF, ADL, ASE, ASG, ASH, ASI, ASJ, ASK, AWA, AWC | PHA, PHB | PVT, AIZ | Cilia, cell bodies, axons | Zwaal et al., 1997; Lans et al., 2004 | ||||
| gpa-4 | ASI | Jansen et al., 1999 | |||||||
| gpa-5 | AWA, ASI, ADL | Cilia, cell bodies, axons, synaptic sites | Jansen et al., 1999; Lans et al., 2004 | ||||||
| gpa-6 | AWA, ASI, AWB, ADL, ASH | PHA, PHB | Dendrites, cell bodies, axons | Jansen et al., 1999; Lans et al., 2004 | |||||
| gpa-7 | ? | ? | Many | Pharyngeal, intestinal, anal sphincter, anal depressor, body wall, vulval | Muscles | Gonad sheath | Jansen et al., 1999 | ||
| gpa-8 | AQR, PQR, URX | Jansen et al., 1999 | |||||||
| gpa-9 | ASJ | PHB | PVQ | Pharyngeal | Muscles | Spermatheca | Jansen et al., 1999 | ||
| gpa-10 | ADF, ASI, ASJ | ALN, CAN, LUA | Spermatheca | Jansen et al., 1999 | |||||
| gpa-11 | ADL, ASH | Jansen et al., 1999 | |||||||
| gpa-12 | Head, ventral cord, tail | Pharyngeal, body wall | Muscles | Hypodermis, intestine, excretory cell | Jansen et al., 1999; Yau et al., 2003; van der Linden et al., 2003 | ||||
| gpa-13 | ADF, ASH, AWC | PHA, PHB | Cilia | Jansen et al., 1999; Lans et al., 2004 | |||||
| gpa-14 | ASI, ASH, ASJ, ASK | ADE, PHA, PHB | ALA, AVA, CAN, DVA, PVQ, RIA | Vulval | Jansen et al., 1999 | ||||
| gpa-15 | ADL, ASH, ASK | PHA, PHB | Male specific | Distal tip cells, anchor cell | Jansen et al., 1999 | ||||
| gpa-16 | AVM, PDE, PLM | BDU, PVC, RIP | Pharyngeal, body wall, vulval | Muscles | Adult gonad | All blastomeres to 4 cell stage, persists in P blastomere and its sister, persists in germ line | Jansen et al., 1999 | ||
| gpa-17 | Intestine | J. Burghoorn and G. Jansen, personal communica- tion | |||||||
| odr-3 | AWA, AWB, AWC, ASH, ADF | PHA or PHB | Cilia, cell bodies, dendrites | Roayaie et al., 1998; Lans et al., 2004 | |||||
| gpb-1 | All | All | All | Body wall | All | Somatic gonad, vulva, hypodermal seam cells, intestine, germ line | Early embryos | Cell membrane, asters before and during early cell divisions | Zwaal et al., 1996 |
| gpb-2 | ? | ? | Most or all | Pharyngeal, body wall, vulval | Muscles | Broadly in head and tail ganglia from comma stage | Outer cell membranes at neuronal cell bodies | van der Linden et al., 2001 | |
| gpc-1 | ADL, ASH, ASJ, AFD, ASI, AWB | PHA, PHB | Jansen et al., 2002 | ||||||
| gpc-2 | All | All | All | All | Jansen et al., 2002 |
Known roles for the Gα subunits in C. elegans are extensive and varied. GOA-1 and GPA-16 function redundantly and are required maternally for proper spindle positioning in the developing embryo (Gotta and Ahringer, 2001; Tsou et al., 2003). GSA-1 and EGL-30 are required for viability after hatching (Brundage et al., 1996; Korswagen et al, 1997). GSA-1, EGL-30 and GOA-1 act both pre- and post-synaptically to affect multiple behaviors and processes. GSA-1 affects locomotion and egg laying, and excess GSA-1 function triggers necrotic neuronal cell death (Korswagen et al., 1997; Berger et al., 1998; Korswagen et al., 1998). EGL-30 activates egg laying, locomotion, pharyngeal pumping, neuronal migration, spicule protraction, and may affect vulval development (Trent et al., 1983; Brundage et al., 1996; Lackner et al., 1999; Miller et al., 1999; Garcia et al., 2001; Kindt et al., 2002; Moghal et al., 2003). GOA-1 negatively regulates locomotion and egg laying, and affects fertility, neuronal migration, pharyngeal pumping, male mating, response to volatile anesthetics, and may also affect vulval development (Mendel et al., 1995; Ségalat et al., 1995; Fraser et al., 2000; Miller and Rand, 2000; Sawin et al., 2000; van Swinderen et al., 2001; Kindt et al., 2002; Keane and Avery 2003). In motor neurons, GSA-1, EGL-30 and GOA-1 act in a network to regulate acetylcholine release. The EGL-30 pathway generates the core signals for synaptic vesicle priming, while the GOA-1 pathway acts to negatively regulate EGL-30 pathway (Lackner et al., 1999; Miller et al., 1999; Nurrish et al., 1999). The GSA-1 pathway acts to modify the EGL-30 pathway, perhaps by providing positional information (Reynolds et al., 2005). Constitutive GPA-12 function affects pharyngeal pumping (van der Linden et al., 2003). Several of the C. elegans specific Gα subunits (GPAs) are involved either positively or negatively in chemosensation (e.g., Roayaie et al., 1998; Jansen et al., 1999).
Only one of the Gβ subunits, GPB-1, appears to function along with the α subunits in the classic G protein heterotrimer (van der Voorn et al., 1990). The remaining Gβ subunit, GPB-2, is thought to promote the GTP-ase activity of Gα subunits (Chase et al., 2001; Robatzek et al., 2001; van der Linden et al., 2001). Of the two Gγ subunits, GPC-2 is broadly expressed and presumably participates in most G protein pathways in C. elegans (Jansen et al., 2002). The remaining Gγ subunit, GPC-1, has a restricted role in chemosensation (Jansen et al., 2002; Hilliard et al., 2005).
The expression and function of each G protein subunit are described in detail below.
C. elegans expresses two Gβ subunits encoded by gpb-1 and gpb-2. GPB-1 shares 86% amino acid identity with mammalian Gβ subunits (van der Voorn et al., 1990), and appears to be required to mediate signaling by all Gα subunits. Inactivation of gpb-1 leads to abnormalities early in embryogenesis (Zwaal et al., 1996; see “Receptor-independent pathway” in the Go section). GPB-1 also has roles later in development that were revealed by mosaic rescue of a gpb-1 null mutant (Zwaal et al., 1996). Sterility and abnormalities in the germ line were observed in some adult mosaic animals, suggesting the gpb-1 may also play a role in germ line development. Overexpression of gpb-1 causes lethargic locomotion and delayed egg laying. These later effects are consistent with the expression pattern of gpb-1; expression is seen throughout development in nearly all somatic tissues and in the germ line (Zwaal et al., 1996). Once tissue differentiation occurs, expression is highest in neurons. In larval and adult stages, GPB-1 expression is seen in most or all neurons, the somatic gonad, vulva, and hypodermal seam cells. The intestine, pharynx, and body wall muscles appear to express GPB-1 at lower levels (Zwaal et al., 1996).
gpb-2 encodes the C. elegans ortholog of vertebrate Gβ5, a Gβ subunit of novel function (Chase et al., 2001; Robatzek et al., 2001; van der Linden et al., 2001). GPB-2 binds G protein γ-like (GGL) containing RGS proteins, and is believed to promote the GTP-ase activity of Gα subunits (Chase et al., 2001; Robatzek et al., 2001; van der Linden et al., 2001). GPB-2 is a regulator of GOA-1 and EGL-30 signaling (see “Regulators of EGL-30/G protein signaling network”).
Two genes encoding Gγ subunits have been identified in C. elegans, gpc-1 and gpc-2 (Jansen et al., 2002). GPC-1 and GPC-2 show from 22 to 79% amino acid identity to the vertebrate Gγ subunits, but are not clear orthologs of any of the human Gγ subunits (Jansen et al., 2002). GPC-1 is specifically expressed in sensory neurons while GPC-2 shows ubiquitous expression (Jansen et al., 2002).
In accordance with its more general pattern of expression, GPC-2 is required together with GPB-1, GOA-1, and GPA-16 to orient cell division axes in the C. elegans embryo (Gotta and Ahringer, 2001) (see “Receptor- independent pathway” in the Gαo section). gpc-2(RNAi) embryos exhibit defects in spindle orientation that are identical to gpb-1(RNAi) and gpb-1 mutant embryos (Gotta and Ahringer, 2001).
GPC-1 is expressed in 12 cells in the head and two cells in the tail that are identified as putative chemosensory neurons. Although GPC-1 is not essential for the detection of the water soluble attractants, NaCl, NaAc, and NH4Cl, it is essential for adaptation to these tastants (Jansen et al., 2002). In addition, gpc-1 mutants exhibit defects in the adaptation to soluble chemical repellents mediated by the ASH neurons, although they are not defective in the response to any repellents tested except for quinine (Hilliard et al., 2005). gpc-1 mutants display wild-type adaptation to volatile odorants (Jansen et al., 2002). gpc-1 overexpression may reduce locomotion and egg laying, though a null mutation in gpc-1 has no effect on those phenotypes (Jansen et al., 2002).
Many of the Gα subunits have remarkably diverse roles in C. elegans biology, and thus these genes have been identified in many genetic screens. Where not previously identified in forward genetic screens, loss-of-function (lf) mutations have been isolated for all Gα subunit genes (with the exception of gpa-17) using target-selected gene inactivation (Jansen et al., 2002). Gα subunits can be mutated to reduce their intrinsic GTP-ase activity, thus causing constitutive signaling. Such gain-of-function (gf) mutations have been isolated for gsa-1 (Schade et al., 2005) and egl-30 (Bastiani et al., 2003), and have been produced in vitro and expressed as transgenes (QL) for gsa-1, goa-1, egl-30, gpa-1, gpa-2, gpa-3, gpa-12 and odr-3 (see sections on individual Gα subunits for references). In many cases, transgenic overexpression of the wild-type Gα subunit (XS) causes effects similar to the activated transgene (although often not as severe) and reciprocal to the loss-of-function mutation. Thus, loss-of-function phenotypes have been described for each of the C. elegans Gα subunits while gain-of-function phenotypes are known for most. We discuss here the current knowledge of their functions.
GSA-1, encoded by gsa-1, shares over 60% identity with its vertebrate ortholog, Gαs. In vertebrates, Gαs is known to activate adenylyl cyclase to produce cyclic AMP, which both activates Protein Kinase A (PKA), and modulates cyclic nucleotide-gated ion channels (reviewed in Sunahara et al., 1996; Walsh and Van Patten, 1994). Vertebrate Gαs can also activate L-type voltage gated calcium channels in skeletal muscle cells and can inhibit cardiac sodium channels (reviewed in Wickman and Clapham, 1995a; Wickman and Clapham, 1995b). In C. elegans, the adenylyl cyclase ACY-1 appears to be the major effector of GSA-1 for growth and locomotion. A receptor with similarity to the vertebrate 5-HT7 family may couple to GSA-1 based on the observation that expression of SER-7b in COS-7 cells results in a dramatic increase in basal cAMP levels over untransfected cells (Hobson et al., 2003).
In C. elegans GSA-1 is required for viability; loss of GSA-1 function results in L1 arrest (Korswagen et al, 1997). GSA-1 also affects egg laying, locomotion, and necrotic neuronal cell death based on mutational analysis (Korswagen et al., 1997; Berger et al., 1998; Korswagen et al., 1998). Viable animals segregating from mosaically rescued transgenic lines show behavioral defects including sluggish locomotion and delayed egg laying (Korswagen et al., 1997). Gain-of-function mutations in gsa-1, like transgenic overexpression, result in reciprocal phenotypes (Korswagen et al, 1997; Schade et al., 2005). Constitutive activation of transgenic GSA-1 causes necrotic neuronal cell death although similar mutations in chromosomal gsa-1 cause only minimal cell death (Korswagen et al, 1997; Schade et al., 2005). GSA-1 plays an ongoing functional role in regulating movement since gsa-1(QL) transgenes are able to induce hyperactive locomotion even when expressed after adulthood (Schade et al., 2005). Gain-of-function mutation in both gsa-1 and acy-1, which encodes an adenylyl cyclase that is the major effector of GSA-1 for movement (see below), cause hypersensitivity to aldicarb, indicating that the GSA-1 pathway regulates acetycholine release by motor neurons. However, elimination of the neuronal GSA-1 pathway does not affect steady-state levels of neurotransmitter release (Reynolds et al., 2005). The implications for this are discussed further in the “Regulators of EGL-30/G protein signaling network” section. GSA-1 also affects gametogenesis, hermaphrodite genital morphogenesis, growth rate and morphogenesis of the epithelium, as determined in large-scale RNAi assays (Simmer et al., 2003).
GSA-1 is broadly expressed in neurons and muscles from the embryonic stage of development onward. It may also be expressed in intestinal and some epithelial cells (i.e. of the pharynx and vulva; Korswagen et al., 1997; Park et al., 1997). GSA-1 functions both pre- and post-synaptically in regulating movement since expression of an activated form of its effector, ACY-1, from promoters specific for either neurons or muscles causes hyperactive locomotion (Schade et al., 2005). GSA-1 pathway activity is also required in both neurons and muscles for larval growth, based on studies using heterologous promoter elements fused to acy-1 (Reynolds et al., 2005).
Two genetic studies of neurodegeneration have revealed conservation between GSA-1 signaling in C. elegans and the vertebrate Gαs signaling pathway (Korswagen et al., 1997; Berger et al., 1998). One screen employed a transgene with the constitutively active C. elegans gsa-1 gene under the control of the ubiquitously expressed heat-shock promoter (Korswagen et al., 1997) and the other employed a transgene with the constitutively active rat Gαs gene under the control of the glr-1 promoter, expressed in 17 classes of neurons, including interneurons required for locomotion (Berger et al., 1998). In both cases, constitutive activation and overproduction of either C. elegans GSA-1 or rat Gαs induced paralysis and neurodegeneration (Korswagen et al., 1997; Berger et al., 1998). Necrotic cell death was suppressed by mutations in acy-1 (sgs-1) (Korswagen et al., 1997; Berger et al., 1998). Most mutations in acy-1 fail to, or only partially suppress, the body-wall hypercontraction caused by transgenic activated Gαs, and also fail to suppress the hyperactive egg laying caused by overproduction of GSA-1 (Korswagen et al., 1997). acy-1 is expressed in the nervous system and in muscle cells (Berger et al., 1998; Korswagen et al., 1998), and null mutations affect larval viability, larval molting, life span, and pharyngeal pumping while reduction-of-function mutations affect locomotion (Moorman and Plasterk, 2002).
For growth and locomotion rate, ACY-1 appears to be the major effector of GSA-1 since null mutations in acy-1 are epistatic to gsa-1(gf) mutations (Schade et al., 2005). Downstream of ACY-1, the GSA-1 pathway likely converges with the EGL-30 pathway to control synaptic transmission from the neurons that regulate locomotion. RIC-8, a GEF protein, may regulate both GSA-1 and EGL-30 with respect to locomotion (Reynolds et al., 2005; see “Regulators of EGL-30/G protein signaling network” section for further detail). A second adenylyl cyclase gene, acy-2, was found to be essential for larval viability (Korswagen et al., 1998). The terminal phenotype of acy-2(lf) mutants resembles that of gsa-1(lf) mutants and clr-1 mutants (Korswagen et al., 1998), which phenotypically mimic worms after laser ablation of the canal-associated neurons (Kokel et al., 1998). acy-2 is expressed in the CAN cells, and some head ganglia and ventral cord neurons (Korswagen et al., 1998). These results indicate that ACY-2 may be an effector of GSA-1 in the CAN cells. Two other predicted adenylate cyclases, acy-3 and acy-4, have not been characterized genetically, nor have RNAi phenotypes been uncovered.
Gαs-induced cell death occurs via a necrotic rather than an apoptotic pathway, based on both the inability of mutations in ced-3 and ced-4 to suppress the cell death and the observation that the morphology of neuronal degeneration observed is similar to that caused by activating mutations in some ion channel genes, such as deg-1, deg-3, and mec-4. Loss-of-function mutations in unc-36, one of three calcium channel genes potentially regulated by PKA (unc-2, unc-36, and egl-19), were found to confer a slight reduction in cytotoxity in one study and slight suppression of paralysis in another (Korswagen et al., 1997; Berger et al., 1998). Cytotoxic cell death mechanisms conferred by activated Gαs and by mec-4(d), deg-1(d) and deg-3(d) alleles require ER-driven Ca2+ release (Xu et al., 2001), and converge upon calcium-regulated aspartyl and calpain proteases (Syntichaki et al., 2002). In all cases, degeneration is suppressed in the genetic mutants cad-1, daf-4, and unc-52, or by starvation, which all reduce aspartyl protease activity. Treatment of mec-4(d), deg-1(d), deg-3(d) and gsa-1(gf) mutant animals with Z-Val-Phe-CHO (MDL-28170), a potent calpain inhibitor, significantly reduces degeneration in these mutants (Syntichaki et al., 2002). These studies suggest that an increase in internal calcium triggers the activation of calpain proteases that subsequently engages executioner lysosomal and cytoplasmic aspartyl proteases and ultimately leads to necrotic cell death (Syntichaki et al., 2002).
egl-30 encodes the ortholog of vertebrate Gαq, and shares over 80% amino acid sequence identity with vertebrate Gαq and Gα11 (Brundage et al., 1996). EGL-30 activates diverse biological processes in C. elegans including egg laying, locomotion, pharyngeal pumping, synaptic transmission, neuronal migration, and spicule protraction in males, and may also promote vulval induction in hermaphrodites (Trent et al., 1983; Brundage et al., 1996; Lackner et al., 1999; Miller et al., 1999; Garcia et al., 2001; Kindt et al., 2002; Moghal et al., 2003). Like vertebrate Gαq family members, EGL-30 has been shown to stimulate phosphoinositide hydrolysis when expressed in COS-7 cells and can couple to the vertebrate α1-C adrenergic receptor (Brundage et al., 1996). EGL-30 has been shown to mediate the release of acetylcholine from motor neurons (Lackner et al., 1999) and is required for serotonin-induced calcium transients in vulval muscles (Shyn et al., 2003).
A complete loss-of-function mutation of egl-30 is presumably inviable (Brundage et al., 1996). Homozygotes having strong reduction-of-function mutations in egl-30 hatch, but are paralyzed with, at most, feeble contractions of body-wall, pharyngeal, and defecation muscles and arrest throughout larval development (Brundage et al. 1996). Less severe alleles cause a variety of phenotypes including sluggish movement, delayed egg laying, slow pharyngeal pumping (Trent et al., 1983; Brundage et al., 1996), and aldicarb resistance (Miller et al., 1999). Gain-of-function mutations in egl-30 or transgenic overexpression of wild type egl-30 cause hyperactive movement with exaggerated body bends, and hyperactive egg laying (Brundage et al., 1996; Bastiani et al., 2003). Overexpression of a constitutively activated form of EGL-30 results in vacuolization of cells, paralysis, and ultimately death (Bastiani et al., 2003). Both loss and gain-of-function mutations in egl-30 cause misplacement of certain migrating neural cell bodies (Kindt et al., 2002).
egl-30 is highly expressed in neurons and in pharyngeal muscle (Lackner et al., 1999; Bastiani et al., 2003). Rescuing fusions with GFP have revealed occasional expression in some other muscle types (i.e., body-wall, vulval) and in some epidermal cells (i.e., vulval; Bastiani et al., 2003). Neuronal expression of egl-30 has been shown to be sufficient for the modulation of locomotion, sensitivity to aldicarb, mediation of retrograde signaling from post-synaptic muscle cells to neurons, and for its effect on vulval development (Lackner et al., 1999; Doi and Iwasaki, 2002; Moghal et al., 2003).
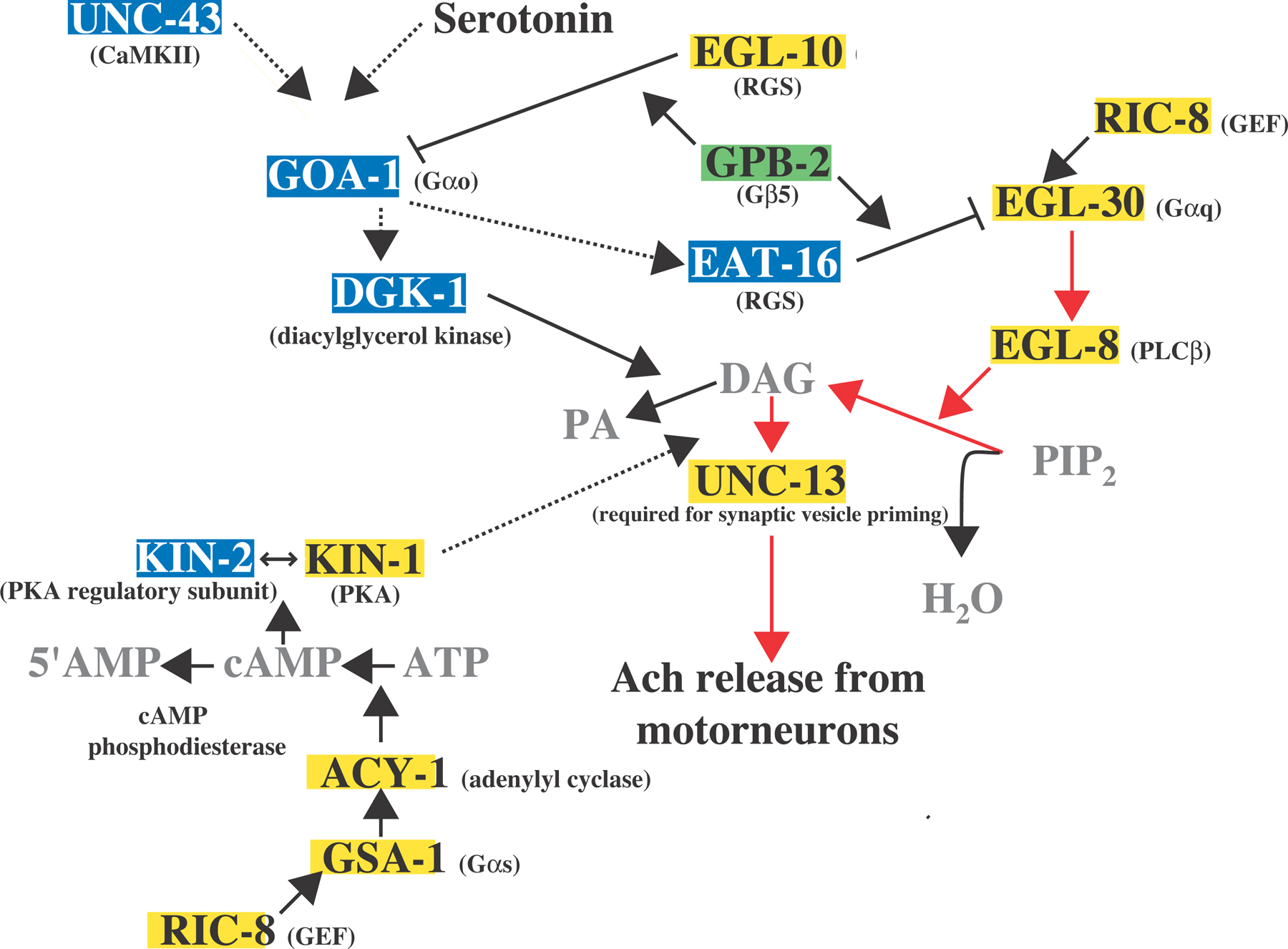
In vertebrates, Gαq activates PLCβ isoforms, which hydrolyze phosphatidylinositol bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (for review, see Sternweis and Smrcka, 1993; Jiang et al., 1994). IP3 interacts with IP3 receptors to stimulate the release of calcium from internal stores while DAG activates PKC. A well-conserved variation of this pathway appears to be employed in C. elegans to mediate acetylcholine release from the ventral cord motor neurons. An increase in the concentration of DAG via activation of EGL-30, and subsequent activation of EGL-8 (PLCβ), is thought to promote presynaptic membrane localization of the DAG receptor protein, UNC-13, a protein that facilitates synaptic vesicle fusion at active zones (Richmond et al., 1999; Richmond et al., 2001). This model is supported by the following observations. Reduction-of-function mutations in egl-30, egl-8, and unc-13 confer reduced sensitivity to aldicarb, an inhibitor of acetylcholinesterase at synapses (Miller et al., 1996; Lackner et al., 1999; Miller et al., 1999). Loss-of-function mutations in dgk-1 (diacylglycerol kinase) can partially bypass the resistance of egl-8 mutants for aldicarb, indicating that acetylcholine release is dependent on DAG levels (Lackner et al., 1999). Both expression of a constitutively active mutation of egl-30 and exposure to phorbol esters cause hyperactive movement and hypersensitivity to aldicarb (Lackner et al., 1999; Reynolds et al., 2005). The effects of increased DAG levels, whether by egl-30(gf) mutations or phorbol ester treatment, are strongly dependent on UNC-13 activity (Lackner et al., 1999; Reynolds et al., 2005). Stimulation of acetylcholine release by phorbol esters, as measured by sensitivity to aldicarb, is blocked by a mutation that eliminates phorbol ester binding to UNC-13 (Lackner et al., 1999). An unc-13(rf) mutation is epistatic to an egl-30(gf) mutation with respect to locomotion. (Reynolds et al., 2005). Expression of an UNC-13 mutant protein that is constitutively membrane-bound restores acetylcholine release to mutants lacking EGL-8 (Lackner et al., 1999). Also, the larval arrest and paralysis of egl-30 null mutants are rescued by phorbol ester treatment (Reynolds et al., 2005). Exogenous treatment of wild-type animals with arecoline, believed to act as an agonist for receptors that are coupled to EGL-30, leads to both hypersensitivity to aldicarb and an increase in the accumulation of UNC-13 at presynaptic membranes (Lackner et al., 1999). Taken together, these data indicate that the pre-synaptic EGL-30 pathway results in synaptic vesicle priming leading to acetylcholine release. This pathway is inhibited by serotonin, via the heterotrimeric Gα protein, GOA-1 (discussed in further detail below; Nurrish et al., 1999).
A number of other processes may also be linked to egl-30-mediated release of acetylcholine. Genetic epistasis with mutations in egl-8 and unc-13 indicate that these genes act downstream of egl-30 with respect to the stimulation of pharyngeal pumping. In this case, itr-1, which is broadly expressed in the pharynx and encodes the C. elegans IP3 receptor, may also act downstream of, or in parallel to, egl-30 (Bastiani et al., 2003). Similarly, an analysis of response to emodepside, a ligand of nematode latrophilin that stimulates neuronal exocytosis and elicits pharyngeal paralysis revealed that latrophilin-dependent neurotransmitter release required egl-30, egl-8 and unc-13 (Willson et al., 2004). Mutations in genes that increase acetylcholine release, such as in unc-43, dgk-1, and egl-8, enhance ectopic axon branching induced by the amino-terminal domain of UNC-6 (Wang and Wadsworth, 2002). egl-30 gain-of-function, as well as reduction-of-function mutants, display defects in neuronal cell migration. Neuronal cell migration is also affected by mutations in egl-8 and dgk-1, although genetic epistasis analysis has not yet been performed (Kindt et al., 2002).
EGL-30 must directly activate other downstream effector molecules in addition to EGL-8, and also mediates and affects other processes in addition to acetylcholine release. The pathway for serotonin-mediated stimulation of egg laying via egl-30 has been shown to be largely independent of EGL-8, thereby specifically implicating another unidentified major effector for EGL-30 (Bastiani et al., 2003). During protraction of the male spicules, egl-30 both enhances a nicotinic pathway and may mediate a muscarinic pathway to affect spicule protraction, and ultimately regulates downstream UNC-68 ryanodine or EGL-19-containing calcium channels (Garcia et al., 2001). EGL-30 activates vulval induction in an EGL-19-dependent manner under specific growth conditions (Moghal et al., 2003). The mechanism by which EGL-30 regulates these channels is not yet known. Further analysis in C. elegans should reveal other direct effectors for EGL-30, and reveal how EGL-30 interacts with other signaling pathways.
EGL-30 may be negatively regulated by the RGS protein EAT-16, as suggested by epistatic relationships between their two genes (Hajdu-Cronin et al., 1999). eat-16(rf) mutations suppress reduction-of-function but not null mutations in egl-30, suggesting that EAT-16 may directly regulate EGL-30 in vivo. Overexpression of EAT-16 partially suppresses phenotypes due to overexpression of EGL-30, but does not suppress those caused by a GTPase deficient EGL-30 transgene (Hajdu-Cronin et al., 1999). In addition, EAT-16 can inhibit EGL-30-dependent phosphoinositide hydrolysis in cultured cells (Hajdu-Cronin et al., 1999).
ric-8 encodes a GEF expressed in neurons that affects egg laying and locomotion. RIC-8 acts genetically upstream of the EGL-30 signaling pathway (Miller and Rand, 2000). Like egl-30(rf) mutations, ric-8(rf) mutations confer aldicarb resistance, suggesting that RIC-8 may act as a positive regulator of EGL-30 for acetylcholine release (Miller et al., 1996; Miller and Rand, 2000). However, although gain-of-function mutations in egl-30 suppress the paralysis of ric-8(rf) mutants, even a strong gain-of-function mutation in egl-30 is unable to suppress a ric-8 null mutation (Reynolds et al., 2005; Schade et al., 2005). An in vitro biochemical analysis of vertebrate Ric-8 indicates that this protein can interact with, and act as a GEF for, multiple Gα subunits (Tall et al., 2003). Similarly, C. elegans RIC-8 can interact with or stimulate the GEF activity in vitro of at least two Gα subunits, GOA-1 and GPA-16 (Afshar et al., 2004; Couwenbergs et al., 2004, Hess et al., 2004). Further studies are needed to address the in vivo specificity of this GEF and its role in EGL-30 signaling.
EGL-30 and GOA-1, the C. elegans ortholog of vertebrate Gαo, confer opposite effects on a variety of behaviors in C. elegans. goa-1 null mutants are hyperactive with respect to locomotion and egg laying whereas egl-30 reduction-of-function mutants are lethargic with respect to these behaviors (Trent et al., 1983; Mendel et al., 1995; Ségalat et al., 1995; Brundage et al., 1996). goa-1 mutants are hypersensitive to aldicarb-induced paralysis and growth arrest, whereas egl-30 mutants are resistant to aldicarb (Miller et al., 1996; Lackner et al., 1999; Miller et al., 1999; Nurrish et al., 1999; van Swinderen et al., 2001).
The observation that goa-1(lf) mutants are resistant to serotonin-induced slowing of locomotion led to the proposal that serotonin and GOA-1 mediate a neuromodulatory effect on acetylcholine release from the ventral cord motorneurons. Indeed, serotonin induces resistance to aldicarb and requires goa-1 to mediate this effect (Nurrish et al., 1999). GOA-1 also negatively regulates the localization of UNC-13 (Nurrish et al., 1999). Mutations in egl-30, and mutations in genes that function in the EGL-30 pathway, are epistatic to mutations in goa-1 with respect to aldicarb sensitivity, suggesting that activation of GOA-1 by serotonin negatively regulates egl-30-mediated acetylcholine release via UNC-13 (Lackner et al., 1999; Miller et al., 1999; Nurrish et al., 1999; for further detail, see the “Pathways” subheading in the EGL-30 section). These pathways appear to intersect at the level of the RGS protein EAT-16, shown to negatively regulate EGL-30, and/or at the level of diacylglycerol kinase, DGK-1, which affects accumulation of DAG (see Figure 2). DGK-1 or EAT-16 might be effectors of GOA-1 since loss-of-function mutations in eat-16 and dgk-1 were isolated in a screen for suppressors of the lethargic and egg-laying defective phenotypes conferred by a goa-1(QL) transgene (Hajdu-Cronin et al., 1999; Miller et al., 1999; Nurrish et al., 1999). Genetic analysis suggests that this signaling network regulates both locomotion and egg laying (Hajdu-Cronin et al., 1999; Miller et al., 1999; Nurrish et al., 1999). It is also likely that unc-43 may act upstream of this signaling network to regulate goa-1 (Robatzek and Thomas, 2000).
GPB-2, the C. elegans ortholog of vertebrate Gβ5, has been shown to regulate the EGL-30/GOA-1 signaling network (Chase et al., 2001; Robatzek et al., 2001; van der Linden et al., 2001). Mutations in gpb-2 were isolated together with mutations in eat-16, goa-1, and dgk-1 as suppressors of unc-43(gf) (Robatzek and Thomas, 2000). Genetic analysis using a targeted deletion of gpb-2 also indicates that GPB-2 modulates the EGL-30/GOA-1 signaling network (see Figure 2; Chase et al., 2001; van der Linden et al., 2001). Like vertebrate Gβ5, GPB-2 has been shown to interact with both C. elegans GGL-containing RGS proteins in vitro (van der Linden et al., 2001). These RGS proteins, EGL-10 and EAT-16, regulate GOA-1 and EGL-30, respectively. (Koelle and Horvitz, 1996; Hajdu-Cronin et al., 1999). Since single amino acid substitutions within the gpb-2 coding region can confer either hyperactive or lethargic egg laying, it is possible that the mutations preferentially affect interactions of GPB-2 with either EGL-10 or EAT-16 (Robatzek et al., 2001). Putative null alleles of gpb-2 are viable, affect egg laying, pharyngeal pumping, and locomotion, and phenotypically resemble animals lacking both EAT-16 and EGL-10 (Chase et al., 2001; van der Linden et al., 2001). gpb-2 is broadly expressed in neurons and in muscle (Chase et al., 2001; van der Linden et al., 2001), so it could be a general regulator of the GOA-1 and EGL-30 signaling pathways. Signaling mechanisms that drive interaction of GPB-2 with either RGS protein remain to be elucidated.
|
Figure 2. Depicted is the G protein signaling network that controls the release of acetylcholine from the motorneurons that regulate locomotion and egg laying (adapted from Reynolds et al., 2005). Proteins highlighted in yellow activate neurotransmitter release and proteins highlighted in blue inhibit neurotransmitter release. The putative regulatory "switch" protein, GPB-2, is highlighted in green. Solid lines indicate strong support for direct interactions, and dotted lines indicate a genetic pathway for which there may be intermediate components. The major stimulatory pathway is the Gαq pathway, indicated by the red arrows. The Gαo pathway inhibits the Gαq pathway by inhibiting presynaptic localization of UNC-13, required for synaptic vesicle priming. DGK-1 and EAT-16 are proposed effectors of GOA-1, but there are no biochemical data to support these models yet. GPB-2 has been shown to interact with both EAT-16 and EGL-10 in vitro, and GPB-2 is believed to stimulate the GAP function of these RGS proteins. Functional Gαq and Gαs pathways are required for locomotion, and the Gαs pathway acts downstream of DAG. Support for this model is based on the following studies: (Brundage et al., 1996; Hajdu-Cronin et al., 1999; Lackner et al., 1999; Miller et al., 1999; Nurrish et al., 1999; Richmond et al., 1999; Robatzek and Thomas, 2000; Chase et al., 2001; Richmond et al., 2001; Robatzek et al., 2001; van der Linden et al., 2001; Reynolds et al., 2005; Schade et al., 2005).
In a screen for suppressors of paralysis caused by ric-8(rf) mutations, Schade et al. (2005) recovered gain-of-function alleles of egl-30, gsa-1, and acy-1 as well as loss-of-function alleles of kin-2, which encodes the regulatory subunit of PKA. Epistasis results using a ric-8 null mutation suggest that RIC-8 is a regulator of both the EGL-30 and GSA-1 pathways in neurons (Reynolds et al., 2005). It has not yet been demonstrated whether RIC-8 can function as a GEF for either of these Gα subunits.
EGL-30 is implicated as a critical component of the core synaptic vesicle priming pathway. On the other hand, GSA-1 does not appear to affect steady-state neurotransmitter release, based on the essentially normal aldicarb sensitivity of an acy-1 null mutant. (Reynolds et al., 2005). Although these proteins do not appear to affect the same process, their pathways appear to merge downstream of DAG (see Figure 2). Either hyperactivation of the EGL-30 pathway or treatment with phorbol esters can rescue acy-1 null mutants with respect to paralysis. Hyperactivation of the GSA-1 pathway in ric-8 null mutants, in combination with phorbol ester treatment, restores locomotion. The authors propose that neuronal GSA-1 is required downstream of DAG for the EGL-30-dependent priming pathway to drive locomotion (Reynolds et al., 2005).
In summary, EGL-30 appears to be negatively regulated by the RGS protein EAT-16 for movement and egg laying (Hajdu-Cronin et al., 1999). In motor neurons, the EGL-30 pathway acts to generate the core signals for synaptic vesicle priming. This pathway is positively regulated by the GEF protein RIC-8 and is negatively regulated by the GOA-1 pathway (Lackner et al., 1999; Miller et al., 1999; Nurrish et al., 1999; Miller and Rand, 2000). The GSA-1 pathway may modify the EGL-30 pathway in motor neurons, perhaps by providing positional information to stabilize vesicle priming at selected synapses optimal for coordinated locomotion (see Figure 2; Reynolds et al., 2005).
In C. elegans, goa-1 encodes the only clear member of the mammalian Gi/o class of Gα subunits. In mammals, Gi/o subunits transduce signals from several hormones and neurotransmitters including acetylcholine, dopamine and serotonin (Neves et al., 2002). The predicted amino acid sequence of C. elegans GOA-1 is over 80% identical to that of mammalian Gαo (Lochrie et al., 1991). Characterization of goa-1 mutants and intensive RNAi analysis has revealed that Gαo signaling in C. elegans regulates several fundamental processes including cell division, neuronal migration, and synaptic transmission. Large-scale RNAi screens have also revealed roles for GOA-1 in egg laying, vulval development, embryonic viability, locomotion, fertility, and the regulation of spindle orientation (Fraser et al., 2000; Simmer et al., 2003).
Null mutations in goa-1 are mostly viable but result in a wide variety of phenotypes including a low frequency (11%) of embryonic lethality (Miller and Rand, 2000), partial sterility (Mendel et al., 1995; Ségalat et al., 1995; Miller and Rand, 2000), defects in neuronal migration (Kindt et al., 2002), and increased levels of acetylcholine release from motor neurons (Miller et al., 1999; Nurrish et al., 1999). goa-1(lf) mutants display a number of behavioral defects including hyperactive locomotion and egg-laying, slow pharyngeal pumping (Mendel et al.,1995; Ségalat et al., 1995), defects in sensory modulation of locomotion and pharyngeal pumping (Sawin et al., 2000; Keane and Avery 2003), and specific defects in male mating (Mendel et al.,1995). goa-1(lf) mutants are also resistant to the effects of volatile anesthetics (van Swinderen et al., 2001). goa-1(XS) or (QL) transgenes generally cause phenotypes opposite to those seen in (lf) mutants (Mendel et al., 1995; Ségalat et al., 1995).
In larval and adult C. elegans, goa-1-reporter gene fusions are expressed throughout the nervous system and in a few non-neuronal cell types including pharyngeal, enteric, and sex-specific muscles, and the hermaphrodite distal tip cells (Mendel et al., 1995; Ségalat et al., 1995). The neuronal expression of GOA-1 has been confirmed by immunolocalization (Miller and Rand, 2000).
In embryos, GOA-1 is present at the cell membrane in all cells (up to at least the 20-cell stage). In the early embryo, GOA-1 is also localized to the cell cortex and to regions surrounding the centrosomal asters (Miller and Rand, 2000; Gotta and Ahringer, 2001).
In neuronal migration and synaptic transmission GOA-1 signaling is mediated by its GTP-bound form, is positively regulated by ligand-activated receptors, and is negatively regulated primarily by the RGS protein EGL-10 (see “Receptor-mediated pathway” below). In early embryonic cleavages, and perhaps in many mitotic cell divisions, GOA-1 function is partially redundant with that of a structurally similar Gα subunit, GPA-16. In this pathway, Gα signaling is receptor independent, negatively regulated by RGS-7, and positively regulated by two nearly identical GPR proteins, GPR-1 and/or GPR-2, and by the GEF protein RIC-8 (see “Receptor-independent pathway” below).
GOA-1 appears to negatively regulate synaptic transmission because goa-1(lf) mutants exhibit hyperactive locomotion and egg laying, and are hypersensitive to aldicarb (Mendel et al., 1995; Ségalat et al., 1995; Miller et al., 1999; Nurrish et al., 1999). Although immediate downstream effectors for GOA-1 have not been definitively identified, GOA-1 likely affects locomotion rate by negatively regulating the acetylcholine release pathway mediated by EGL-30 (discussed under “Negative regulation of the EGL-30 pathway by GOA-1”). The role of GOA-1 in egg laying is likely complex since it acts both pre- and post-synaptically (Mendel et al., 1995; Nurrish et al., 1999; Shyn et al., 2003). Presynaptically, GOA-1 may function in egg laying as it does in the ventral cord motor neurons; inhibiting neurotransmitter release by negatively regulating the EGL-30 pathway (Bany et al., 2003). Postsynaptically, GOA-1 appears to negatively regulate vulva muscle activity independent of input from the egg-laying motor neurons (Shyn et al., 2003).
For locomotion and egg laying, GOA-1 is negatively regulated by the RGS protein EGL-10. Mutations in egl-10 cause phenotypes opposite to those seen in goa-1, and goa-1(lf) mutations are epistatic to those in egl-10 (Koelle and Horvitz, 1996). Two other RGS proteins, encoded by rgs-1 and rgs-2, can also act as GTPase activators for GOA-1 (Dong et al., 2000). Loss-of-function mutations in rgs-1 or rgs-2 are not opposite to those of goa-1. Instead, RGS-1 and RGS-2 are redundantly required to rapidly induce egg laying when starved animals are re-fed (Dong et al., 2000).
The signaling molecules that directly activate a GOA-1-coupled receptor have not yet been defined. However, several neurotransmitters and neuromodulators appear to act upstream of GOA-1. goa-1(lf) mutants are partially resistant to the effects of serotonin on locomotion, egg-laying and defecation, suggesting that GOA-1 may act as a serotonin effector (Mendel et al., 1995; Ségalat et al., 1995). Exogenous serotonin reduces locomotion rate by reducing acetylcholine release at neuromuscular junctions, resulting in resistance to aldicarb. This effect requires GOA-1 activity (Nurrish et al., 1999). Although expression of goa-1 in ventral cord motor neurons is sufficient to alter aldicarb responsiveness, the site of action of serotonin’s effect on movement has not been established (Nurrish et al., 1999). Serotonin also acts upstream of GOA-1 in a pathway that mediates the enhanced slowing response seen when starved worms are reintroduced to food (Sawin et al., 2000; Ranganathan et al., 2001). Additionally, spontaneous activity of the egg-laying motorneurons is silenced by serotonin through a pathway that requires GOA-1 activity (Shyn et al., 2003). GOA-1 also acts downstream of serotonin in the migrating neurons ALM L/R, BDU L/R, SDQR and AVM, perhaps transducing the serotonin signal to the UNC-2 calcium channel (Kindt et al., 2002). In addition to serotonin, other neurotransmitters act upstream of GOA-1, including FRMFamides (Nelson et al., 1998; Rogers et al., 2001), acetylcholine (Bany et al., 2003), octopamine (Rogers et al., 2001) and dopamine (Sawin et al., 2000). GOA-1 may also transduce a signal present in a dauer pheromone preparation that confers resistance to volatile anesthetics (van Swinderen et al., 2002).
Loss-of-function mutations in goa-1 cause a low percentage of embryonic lethality (Miller and Rand, 2000). Strong synthetic embryonic lethality is seen when both GOA-1 and a structurally similar Gα subunit, GPA-16, are depleted, either by RNA interference (RNAi) or mutation (Gotta and Ahringer, 2001; Bergmann et al., 2003). Depletion of both Gα subunits causes multiple defects in cell cleavage in the early embryo. During the first mitotic cleavage, nuclear rotation does not occur, spindle movement to the posterior is reduced or absent, and the posterior spindle aster remains spherical and fails to oscillate (Gotta and Ahringer, 2001; Tsou et al., 2003). GOA-1 and GPA-16 are also required to generate most or all of the pulling force on spindle poles during mitosis (Colombo et al., 2003). Although first cleavage is nearly symmetric in embryos depleted for GOA-1 and GPA-16, other asymmetries in the zygote (P0) appear normal, suggesting that the Gα subunits act downstream of or in parallel to the PAR proteins (Gotta and Ahringer, 2001). Following the first cleavage in GOA-1/GPA-16 depleted embryos, nuclei are mispositioned and nuclear rotation fails to occur in the blastomere P1 (Gotta and Ahringer, 2001; Tsou et al., 2003). During the next rounds of cell division, centrosomes fail to separate and spindle orientations are incorrect (Gotta and Ahringer, 2001). Embryos arrest with polyploid nuclei and few cells (Srinivasan et al., 2003).
In the early embryo, GOA-1 and GPA-16 partner with the Gβ subunit GPB-1 and the Gγ subunit GPC-2 (Gotta and Ahringer, 2001). It had previously been proposed that some of the aberrations in early cleavage are due to Gβγ signaling (Gotta and Ahringer, 2001). However, potent depletion of GOA-1/GPA-16 is epistatic to depletion of GPB-1 or GPC-2, indicating that regulation of spindle positioning and nuclear rotation in the early embryo acts via Gα signaling (Tsou et al., 2003). The G proteins, as well as their regulators described below, are required maternally to promote normal embryonic development (Zwaal et al., 1996; Miller and Rand, 2000; Srinivasan et al., 2003).
Additional proteins have been identified that function along with GOA-1 and GPA-16 in the embryo. ric-8(lf) mutations result in the same embryonic phenotypes seen when both GOA-1 and GPA-16 are depleted (Miller and Rand, 2000; Afshar et al., 2004; Couwenbergs et al., 2004). RIC-8 can physically interact with both GOA-1 and GPA-16 and has been shown to act as a GEF for GOA-1 (Afshar et al., 2004; Couwenbergs et al., 2004).
Depletion of GPR-1 and GPR-2, two nearly identical homologs of the receptor-independent G protein regulator AGS3/PINS, also causes the same repertoire of early cleavage defects (Colombo et al., 2003; Gotta et al., 2003; Srinivasan et al., 2003). GPR-1/2 binds to the GDP-bound form of GOA-1 and acts as a GDP dissociation inhibitor (GDI; Colombo et al., 2003; Gotta et al., 2003; Afshar et al., 2004). LIN-5 is also required for correct spindle orientation and functions to anchor and localize GPR-1/2 (Gotta et al., 2003; Srinivasan et al., 2003).
One RGS protein, RGS-7, is required for embryonic viability (Hess et al., 2004). In rgs-7(lf) mutants, the posterior centrosomes show exaggerated movements compared to wild-type, and the force pulling on the anterior spindle pole is reduced (Hess et al., 2004). Depletion of GOA-1/GPA-16, GPR-1/2 or RIC-8 is epistatic to loss of rgs-7 function, indicating that the effects of RGS-7 on spindle positioning require Gα and its regulators (Hess et al., 2004). The effects of RGS-7 on spindle force are asymmetric although the expression of rgs-7 appears symmetric (Hess et al., 2004). It is possible that RGS-7 functions in concert with the asymmetrically distributed LET-99 protein. LET-99 contains a DEP domain, as do certain RGS proteins including EGL-10, the RGS that regulates GOA-1 activity in adult neurons. Both the DEP and RGS domains are required for EGL-10 function, and these domains can regulate GOA-1 when expressed on separate peptides (Patikoglou and Koelle, 2002). Like RGS-7, LET-99 functions antagonistically to GOA-1/GPA-16 and GPR-1/2, and loss-of-function mutations in let-99 are hypostatic to depletion of GOA-1/GPA-16, GPR-1/2, or RIC-8 (Tsou et al., 2003; Couwenbergs et al., 2004).
Several models describing the receptor-independent G protein cycle involved in spindle force have been proposed. Since both GOA-1 and GPR-1/2 act positively to regulate cell cleavage, it has been proposed that the signaling molecule is a Gα-GDP/GPR complex (Colombo et al., 2003; Gotta et al., 2003; Afshar et al., 2004). Alternatively, Gα-GTP produced by the GEF activity of RIC-8 could be the signaling molecule (Gotta et al., 2003; Srinivasan et al., 2003). Many models suggest that the role of GPR-1/2 is to compete with Gβγ for binding to Gα with the resulting Gα-GDP/GPR complex acting as a substrate for RIC-8 activity (Gotta et al., 2003; Srinivasan et al., 2003; Couwenbergs et al., 2004; Hess et al., 2004). However, RIC-8 appears to act prior to GPR-1/2 since RIC-8 is required for the association of GPR-1/2 with GOA-1, but GPR-1/2 is not required for the association of RIC-8 with GOA-1 (Afshar et al., 2004). At present none of the proposed models encompass all the available data. Moreover, much of the in vitro analysis has not included GPA-16, whose role might not be identical to that of GOA-1. Determination of the active signaling molecule or molecules will be critical to predicting and interpreting epistatic interactions among components of this pathway.
gpa-12 encodes a Gα12/13 ortholog with 52% identity to human Gα12. Mouse Gα12 may interact with the Gαq and Gα13 pathways to affect embryonic development (Gu et al., 2002). Vertebrate Gα12 is capable of transforming mouse fibroblast cell lines (Chan et al., 1993; Voyno-Yasenetskaya et al., 1994) and promotes stress fiber formation in cultured fibroblast cells (Buhl et al., 1995; Fromm et al., 1997; Katoh et al., 1998; Gohla et al., 1999; Lowry et al., 2002). Gα12 is linked with downstream activation of Rho and with activation of the serum response promoter element (SRE) in the c-fos protooncogene (Buhl et al., 1995; Fromm et al., 1997; Katoh et al., 1998).
Analysis of a gpa-12 null mutation as well as a large-scale RNAi analysis, indicate that gpa-12 is not required for viability and that reducing or eliminating its expression does not elicit obvious defects in C. elegans (Kamath et al., 2003; van der Linden et al., 2003). However, another RNAi analysis revealed effects on viability as well as on locomotion, oogenesis, and touch sensitivity (Yau et al., 2003). The reason for these differences is not clear. It is possible that in the latter study, the effects of RNAi may be due to partial silencing of other Gα subunit genes.
Animals that overexpress constitutively activated GPA-12 exhibit developmental growth arrest due to a dramatic reduction in pharyngeal pumping.
gpa-12 is predominantly expressed in the pharynx and in hypodermal cells, and has shown variable expression in some other muscle and intestinal cells, and in a few neurons in the head, ventral cord and tail of the animal (Jansen et al., 1999; van der Linden et al., 2003; Yau et al., 2003).
A genetic screen to recover mutations that suppress the growth arrest phenotype caused by activated GPA-12 revealed one potential downstream target, TPA-1 (van der Linden et al., 2003). TPA-1 is a protein kinase C homolog most similar to the vertebrate novel calcium-independent PKCθ/δ (van der Linden et al., 2003). The site-of-action for growth arrest mediated by GPA-12, as well as suppression mediated by TPA-1, was shown to be in the pharyngeal muscle, based on heterologous promoter fusions with myo-2 (van der Linden et al., 2003), suggesting that these proteins may directly interact with one another.
Activation of Rho by Gα12/13 has been shown, in some cases, to occur specifically through interaction of these Gα proteins with RhoGEFs (Kozasa et al., 1998; Suzuki et al., 2003; Dutt et al., 2004). A CeRhoGEF homolog has been shown to interact with GPA-12 when these proteins are coexpressed in COS-7 cells (Yau et al., 2003). The CeRhoGEF, in turn, has been shown to stimulate serum response factor (SRF) activation, and this activation is inhibited by C3 botulinus toxin, which specifically inactivates Rho by ADP ribosylation. While CeRhoGEF expression appears primarily neuronal, at most weak expression of GPA-12 is observed in neurons, so this pathway may be conserved only in specific tissues in C. elegans. RNAi analysis of CeRhoGEF indicates that it plays a role in embryonic development (Yau et al., 2003).
There are 17 Gα subunits, gpa-1-11, gpa-13-17, and odr-3 that are not clear orthologs of mammalian proteins, although they most closely resemble the Go/i class (Fino Silva and Plasterk, 1990; Lochrie et al., 1991; Roayaie et al., 1998; Jansen et al., 1999; Cuppen et al., 2003).
For most of the gpa genes, expression is confined to small subsets of mostly sensory neurons. The exceptions are: gpa-7, expressed in most neurons and in all muscle cells, gpa-14, expressed in sensory and a few other neurons and in the vulva muscles, gpa-15, expressed in the distal tip cell and anchor cell as well as sensory neurons, gpa-16, expressed in adults in several neurons, the pharynx and in body wall and vulva muscles, and gpa-17, expressed in the intestine (Jansen et al., 1999; J. Burghoorn and G. Jansen, personal communication). gpa-1, gpa-7 and gpa-15 are also expressed in male-specific neurons (Jansen et al., 1999). See Table 1.
The function of each gpa gene has been investigated by examining both loss-of-function (lf) mutants, most obtained by target-selected gene inactivation, and gain-of-function animals overexpressing either a wild type (XS) or constitutively activated (QL) transgene (Zwaal et al., 1997; Roayaie et al., 1998; Jansen et al., 1999). odr-3(lf) mutants were isolated in screens for mutants defective in osmotic avoidance and in chemotaxis to benzaldehyde (Roayaie et al., 1998). Phenotypes caused by mutations in each gpa gene, where known, are described briefly below. Negative results are also included to indicate which phenotypes have been examined. Since many of the GPA proteins function redundantly, interactions between them are described under ‘Pathways’ below.
gpa-1(lf) animals are wild type for response to both soluble and volatile attractants and repellents. gpa-1(XS) transgenics are defective for response to soluble attractants and repellents, and males have reduced potency (Jansen et al., 1999). gpa-1(QL) animals have no visible phenotype and males have apparently normal mating behavior (Jane Mendel, unpublished observations).
gpa-2(lf) mutants are wild type for response to both soluble and volatile attractants and repellents (Roayaie et al., 1998; Jansen et al.,1999). Roayaie et al. (1998), reported that gpa-2(lf) mutations enhance the mild defect in chemotaxis to butanone seen in odr-3(lf) mutants, indicating that GPA-2 and ODR-3 act redundantly in response to this odorant. However, Lans et al., (2004), found improved odorant responses in gpa-2(lf)odr-3(lf) double mutants for all odorants except isoamylalcohol and butanone, indicating a negative function for GPA-2 in detection of most odorants. In their study, butanone response was too low in odr-3 mutants to detect a redundant stimulatory function for GPA-2 (Lans et al., 2004). gpa-2(lf) mutants are partially resistant to dauer pheromone with respect to dauer formation and recovery from the dauer stage (Zwaal et al., 1997). gpa-2(XS) transgenics are completely resistant to dauer pheromone, whereas expression of the (QL) transgene causes both constitutive dauer formation and resistance to exogenous pheromone (Zwaal et al., 1997). gpa-2(QL) animals are wild type for response to water-soluble attractant and repellents, but are defective for attraction and aversion to certain volatile odorants. Male potency is not affected by the (QL) transgene (Jansen et al., 1999).
gpa-3(lf) mutants are defective in response to water-soluble attractants and repellents, and are partially resistant to dauer induction by pheromone, but are sensitive to pheromone inhibition of dauer recovery (Zwaal et al., 1997; Jansen et al., 1999; Hilliard et al., 2004). gpa-3(lf) mutants are also defective in response to the volatile attractant pyrazine (Jansen et al., 1999). GPA-3 is sufficient for detection of all odorants except butanone (Lans et al., 2004). Loss of gpa-3 activity enhances the chemotaxis defect of odr-3(lf) mutants to all odorants indicating that GPA-3 is redundant with ODR-3, and that it plays a stimulatory role in odorant detection (Lans et al., 2004). gpa-3(QL) transgenics are defective in response to water-soluble attractants and to several volatile odorants, and like gpa-3(XS) transgenics are resistant to dauer pheromone (Zwaal et al., 1997; Jansen et al., 1999). The underlying basis for the chemosensory and dauer defects in gpa-3(QL) transgenics may be due to defects in morphological development of the amphid sensory neurons since these animals are defective for dye filling of amphid neurons (Zwaal et al., 1997). The gpa-3(QL) transgene causes a Daf-c phenotype (Zwaal et al., 1997).
gpa-4(lf) mutants are wild type for chemosensation while gpa-4(XS) transgenics are defective in attraction to some water-soluble chemicals (Jansen et al., 1999).
gpa-5(lf) mutants are wild type for response to soluble attractants and repellents, but show increased sensitivity to volatile attractants sensed by AWA (Jansen et al., 1999), and to one attractant, isoamylalcohol, sensed by AWC (Battu et al., 2003). gpa-5(lf) mutations rescue the chemotaxis defect of odr-3 mutants to all odorants detected by AWA, suggesting that gpa-5 is a negative regulator of chemosensation in that cell (Jansen et al., 1999; Lans et al., 2004). Additionally, gpa-5(lf) mutations suppress the Vul phenotype of let-60(rf) and sem-5(rf) mutations, suggesting an inhibitory role for GPA-5 in vulval induction (Battu et al., 2003).
gpa-6(lf) and gpa-6(XS) mutants are wild type for chemotaxis to soluble and volatile attractants and repellents. gpa-6(lf) mutants show and increased preference for NaCl when presented with a choice between NaCl and NaAc (Jansen et al., 1999).
gpa-7(lf) mutants are wild type for chemotaxis, except for a defect in aversion to low concentrations of SDS. gpa-7(XS) transgenics are defective for chemotaxis to both water soluble and volatile attractants and repellents (Jansen et al., 1999). gpa-7(lf) mutants are egg-laying defective, while gpa-7(XS) transgenics are hyperactive for this behavior, suggesting that gpa-7 plays a stimulatory role in egg-laying (Jansen et al., 1999). However, gpa-7(lf) mutations have no significant effect on vulva muscle activity (Shyn et al., 2003).
gpa-8(lf) and gpa-8(XS) mutants are wild type for chemotaxis, male potency and dye filling (Jansen et al., 1999).
gpa-9(lf) mutants are wild type for chemotaxis, male potency and dye filling (Jansen et al., 1999).
gpa-10(lf) mutants are wild type for chemotaxis, male potency and dye filling. gpa-10(XS) transgenics are defective for response to water-soluble attractants and repellents, and defective for response to pyrazine and 2-nonanone. Dye filling of amphid neurons is stronger in gpa-10(XS) animals (Jansen et al., 1999).
gpa-11(lf) mutants are wild type for male potency, dye filling and chemotaxis to odorants (Jansen et al., 1999). However, these mutants respond poorly to the repellent octanol (Chao et al., 2004). gpa-11(XS) transgenics are defective in response to glucose and have reduced male potency (Jansen et al., 1999).
gpa-13(lf) mutants are mildly defective for response to 2,3-pentanedione. This phenotype is weakly enhanced by mutations in odr-3, suggesting that GPA-13 plays a minor stimulatory role in olfaction (Lans et al., 2004).
gpa-14(lf) mutants are wild type for chemotaxis, male potency and dye filling (Jansen et al., 1999). Although expressed in vulval muscle, gpa-14(lf) mutations have no significant effect on vulval muscle activity (Shyn et al., 2003).
gpa-15(lf) mutants are wild type for chemotaxis, male potency and dye filling (Jansen et al., 1999).
gpa-16(lf) mutations interact synergistically with goa-1(lf) mutations to affect early cleavages in C. elegans as described above under Gαo “Receptor Independent Pathway”.
gpa-17(RNAi) has no obvious effects (J. Burghoorn and G. Jansen, personal communication; Maeda et al., 2001; Kamath et al., 2003).
odr-3(lf) mutants have reduced responses to all volatile attractants detected by AWA and AWC and are also defective in the chemosensory and mechanosensory functions of ASH (Roayaie et al., 1998; Hilliard et al., 2004). In addition, odr-3(lf) mutants have altered AWC cilia with filamentous branches similar to AWA (Roayaie et al., 1998). odr-3(XS) transgenics are defective in response to all odorants sensed by AWA and AWC, but are not defective in osmotic avoidance. Overexpression of odr-3 causes alterations in the morphology of AWA cilia such that the filamentous branches become more AWC-like. The amount of ODR-3 can thus determine the extent of cilium outgrowth in AWA and AWC (Roayaie et al., 1998). odr-3(QL) transgenics are defective in olfaction and osmotic avoidance and have abnormal AWC cilia (Roayaie et al., 1998).
ASH, which mediates avoidance behavior, expresses multiple Gα subunits (Jansen et al., 1999; see Table 1). Osmotic avoidance, avoidance of nose touch and avoidance of the volatile repellent octanol require ODR-3 (Roayaie et al., 1998). Avoidance of octanol is mediated by serotonin and food and also requires GPA-11 (Chao et al., 2004). Overexpression of odr-3 partially rescues the octanol avoidance defect of grk-2(lf) mutants which lack the G-protein-coupled receptor kinase GRK-2 (Fukuto et al., 2004). ASH also mediates avoidance of the water-soluble repellents quinine, heavy metal ions (copper), and SDS (Sambongi et al., 1999; Hilliard et al., 2004; Hilliard et al., 2005). Response to these repellents, as measured by calcium imaging in ASH, is reduced in odr-3(lf) mutants and abolished in gpa-3 odr-3 double mutants indicating functional redundancy for ODR-3 and GPA-3 in ASH (Hilliard et al., 2005). The gpa-3(lf) mutation alone significantly reduces the response to quinine but not to copper or SDS (Hilliard et al., 2005). GPA-3 also shows partial redundance with ODR-3 in sensing hyperosmolarity (Hilliard et al., 2005).
AWA, which mediates response to volatile odorants including diacetyl, pyrazine and 2,4,5,-trimethylthiazole, expresses four Gα genes, gpa-3, gpa-5, gpa-6, and odr-3 (Roayaie et al., 1998; Jansen et al., 1999; Lans et al., 2004). ODR-3 provides the main stimulatory signal; GPA-3 also provides a stimulatory signal (Lans et al., 2004). GPA-5 has an inhibitory function in AWA, probably via the GPCR SRA-13 (Battu et al., 2003; Lans et al., 2004). Chemotaxis to diacetyl also requires GRK-2. The reduced response to diacetyl seen in grk-2(lf) mutants is significantly restored in mutants lacking the RGS protein EAT-16, suggesting that EAT-16 may downregulate G protein signaling in AWA (Fukuto et al., 2004). Deletion of the inhibitory Gα subunit GPA-5 does not restore chemotaxis in grk-2(lf) mutants (Fukuto et al., 2004).
AWC, which mediates response to volatile attractants including benzaldehyde, butanone, and isoamylalcohol, expresses the Gα genes gpa-2, gpa-3, gpa-13 and odr-3 (Roayaie et al., 1998; Jansen et al., 1999; Lans et al., 2004). As in AWA, ODR-3 provides the main stimulatory signal and GPA-3 also has a stimulatory function (Lans et al., 2004). For many odorants, GPA-2 plays an inhibitory role in AWC, although GPA-2 may also play a redundant stimulatory role in the response to butanone (Roayaie et al., 1998; Lans et al., 2004). Although expression of GPA-5 has not been detected in AWC, gpa-5(lf) mutants are hypersensitive to isoamylalcohol and suppress the reduced response to this odorant caused by overexpression of SRA-13, a GPCR expressed in both AWA and AWC (Battu et al., 2003).
GPA-2 and GPA-3 are involved redundantly, but not exclusively in the dauer decision. Constitutive activation of either protein results in constitutive dauer formation. Both gpa-2(lf) and gpa-3(lf) mutants are partially resistant to dauer induction by pheromone. The gpa-2 gpa-3 double mutant is less sensitive to dauer induction by pheromone than either single mutant indicating functional redundancy. However, the double mutant does form a low percentage of dauers in response to pheromone, revealing an additional pathway involved in pheromone response (Zwaal et al., 1997). In addition to promoting dauer development, pheromone also inhibits recovery of dauer larvae in the presence of food. gpa-3(lf) mutant dauers are sensitive to this inhibition, but gpa-2(lf) dauers are able to recover in the presence of pheromone. gpa-2 is epistatic to gpa-3 for this phenotype. Constitutively activated GPA-2 does not require GPA-3 to promote dauer formation and, similarly, activated GPA-3 does not require GPA-2 (Zwaal et al., 1997). These data are consistent with GPA-2 and GPA-3 acting in parallel to regulate the response to dauer pheromone.
GPA-3 is expressed in the amphid neurons known to be involved in dauer formation, however, GPA-2 is not detectably expressed in those cells (Zwaal et al., 1997; Jansen et al., 1999; Lans et al., 2004). The Daf-c phenotype caused by the activated gpa-2 and gpa-3 transgenes is suppressed by cilium-structure mutations, suggesting that these G proteins function in or via cilia. Immunostaining indicates expression of both GPA-2 and GPA-3 in sensory cilia, although both proteins are also localized to cell bodies and axons (Lans et al., 2004).
The (lf) mutants and the (QL) transgenics for both gpa-2 and gpa-3 show appropriate responses to temperature and food (Zwaal et al., 1997). However, transgenic overexpression of either wild type or activated gpa-2 or gpa-3 causes resistance to pheromone. For gpa-3, this may be because the transgenes cause abnormal amphid neuron morphology (Zwaal et al., 1997; Jansen et al., 1999). However, gpa-2 transgenics have apparently normal amphid cilia but still fail to respond to pheromone. This could be due to activation of negative regulatory pathways or adaptation of downstream components of the signaling cascade.
Genetically, gpa-2 and gpa-3 appear to act downstream of or in parallel to daf-11 and daf-21, and upstream of or in parallel to daf-1 and daf-8 (Zwaal et al., 1997).
GPA-5 acts downstream of the GPCR SRA-13 to negatively regulate vulva development by negatively regulating RAS/MAPK signaling. sra-13(lf) mutations suppress the vulvaless (Vul) phenotype caused by let-60 ras (rf) or (dn) mutations. The sra-13(lf) mutation is not able to suppress stronger Vul phenotypes caused by mutations in sem-5. gpa-5(lf) mutations are more robust and can suppress the Vul phenotype of both let-60(rf) and sem-5(rf) mutations (Battu et al., 2003). GPA-5, therefore, may transduce signals from other GPCRs in addition to SRA-13. Vulval induction is sensitive to food levels, with starvation suppressing the multivulva (Muv) phenotype of let-60(gf) mutants. This suppression requires functional sensory neurons. In sra-13(lf); let-60(gf) double mutants, vulval induction is not reduced by starvation, suggesting that SRA-13 is required to transmit sensory information about food levels to the VPCs. SRA-13 and GPA-5 could act cell autonomously in the VPCs to inhibit vulval development. Alternatively, and more consistent with their detected expression patterns, SRA-13 and GPA-5 could act in sensory neurons to produce a secondary signal that downregulates RAS/MAPK activity (Battu et al., 2003).
Afshar, K., Willard, F.S., Colombo, K., Johnston, C.A., McCudden, C.R., Siderovski, D.P., and Gonczy, P. (2004). RIC-8 is required for GPR-1/2-dependent Gα function during asymmetric division of C. elegans embryos. Cell 119, 219–230. Abstract Article
Bany, I.A., Dong, M.Q., and Koelle, M.R. (2003). Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J. Neurosci. 23, 8060–8069. Abstract
Bastiani, C.A., Gharib, S., Simon, M.I., and Sternberg, P.W. (2003). Caenorhabditis elegans Gαq regulates egg-laying behavior via a PLCβ-independent and serotonin-dependent signaling pathway and likely functions both in the nervous system and in muscle. Genetics 165, 1805–1822. Abstract Article
Battu, G., Hoier, E.F., and Hajnal, A. (2003). The C. elegans G-protein-coupled receptor SRA-13 inhibits RAS/MAPK signalling during olfaction and vulval development. Development 130, 2567–2577. Abstract Article
Berger, A.J., Hart, A.C., and Kaplan, J.M. (1998). Gαs-induced neurodegeneration in Caenorhabditis elegans. J. Neurosci. 18, 2871–2880. Abstract
Bergmann, D.C., Lee, M., Robertson, B., Tsou, M.F., Rose, L.S., and Wood, W.B. (2003). Embryonic handedness choice in C. elegans involves the Gα protein GPA-16. Development 130, 5731–5740. Abstract Article
Betz, A., Ashery, U., Rickmann, M., Augustin, I., Neher, E., Sudhof, T.C., Rettig, J., and Brose, N. (1998). Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron 21, 123–136. Article
Brose, N., Rosenmund, C., and Rettig, J. (2000). Regulation of transmitter release by Unc-13 and its homologues. Curr. Opin. Neurobiol. 10, 303–311. Article
Brundage, L., Avery, L., Katz, A., Kim, U.J., Mendel, J.E., Sternberg, P.W., and Simon, M.I. (1996). Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron 16, 999–1009. Abstract Article
Buhl, A.M., Johnson, N.L., Dhanasekaran, N., and Johnson, G.L. (1995). Gα12 and Gα13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 270, 24631–24634. Abstract Article
Chan, A.M., Fleming, T.P., McGovern, E.S., Chedid, M., Miki, T., and Aaronson, S.A. (1993). Expression cDNA cloning of a transforming gene encoding the wild-type Gα12 gene product. Mol. Cell Biol. 13, 762–768. Abstract
Chao, M.Y., Komatsu, H., Fukuto, H.S., Dionne, H.M., and Hart, A.C. (2004). Feeding status and serotonin rapidly and reversibly modulate a Caenorhabditis elegans chemosensory circuit. Proc. Natl. Acad. Sci. USA 101, 15512–15517. Abstract Article
Chase, D.L., Patikoglou, G.A., and Koelle, M.R. (2001). Two RGS proteins that inhibit Gαo and Gαq signaling in C. elegans neurons require a Gβ5-like subunit for function. Curr. Biol. 11, 222–231. Abstract Article
Colombo, K., Grill, S.W., Kimple, R.J., Willard, F.S., Siderovski, D.P., and Gonczy, P. (2003). Translation of polarity cues into asymmetric spindle positioning in Caenorhabditis elegans embryos. Science 300, 1957–1961. Abstract Article
Couwenbergs, C., Spilker, A.C., and Gotta, M. (2004). Control of embryonic spindle positioning and Gα activity by C. elegans RIC-8. Curr. Biol. 14, 1871–1876. Abstract Article
Cuppen, E., van der Linden, A.M., Jansen, G., and Plasterk, R.H. (2003). Proteins interacting with Caenorhabditis elegans Gα subunits. Comparative and Functional Genomics 4, 479–491. Article
Doi, M., and Iwasaki, K. (2002). Regulation of retrograde signaling at neuromuscular junctions by the novel C2 domain protein AEX-1. Neuron 33, 249–259. Abstract Article
Dong, M.Q., Chase, D., Patikoglou, G.A., and Koelle, M.R. (2000). Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev. 14, 2003–2014. Abstract
Dutt, P., Nguyen, N., and Toksoz, D. (2004). Role of Lbc RhoGEF in Gα12/13-induced signals to Rho GTPase. Cell Signal 16, 201–209. Abstract Article
Fino Silva, I., and Plasterk, R.H. (1990). Characterization of a G-protein α-subunit gene from the nematode Caenorhabditis elegans. J. Mol. Biol. 215, 483–487. Abstract
Fraser, A.G., Kamath, R.S., Zipperlen, P., Martinez-Campos, M., Sohrmann, M., and Ahringer, J. (2000). Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408, 325–330. Abstract Article
Fromm, C., Coso, O.A., Montaner, S., Xu, N., and Gutkind, J.S. (1997). The small GTP-binding protein Rho links G-protein-coupled receptors and Gα12 to the serum response element and to cellular transformation. Proc. Natl. Acad. Sci. USA 94, 10098–10103. Abstract Article
Fukuto, H.S., Ferkey, D.M., Apicella, A.J., Lans, H., Sharmeen, T., Chen, W., Lefkowitz, R.J., Jansen, G., Schafer, W.R., and Hart, A.C. (2004). G-protein-coupled receptor kinase function is essential for chemosensation in C. elegans. Neuron 42, 581–593. Abstract Article
Garcia, L.R., Mehta, P., and Sternberg, P.W. (2001). Regulation of distinct muscle behaviors controls the C. elegans male's copulatory spicules during mating. Cell 107, 777–788. Abstract Article
Gohla, A., Offermanns, S., Wilkie, T.M., and Schultz, G. (1999). Differential involvement of Gα12 and Gα13 in receptor-mediated stress fiber formation. J. Biol. Chem. 274, 17901–17907. Abstract
Gotta, M., and Ahringer, J. (2001). Distinct roles for Gα and Gβγ in regulating spindle position and orientation in Caenorhabditis elegans embryos. Nat. Cell Biol. 3, 297–300. Abstract Article
Gotta, M., Dong, Y., Peterson, Y.K., Lanier, S.M., and Ahringer, J. (2003). Asymmetrically distributed C. elegans homologs of AGS3/PINS control spindle position in the early embryo. Curr. Biol. 13, 1029–1037. Abstract Article
Gu, J.L., Muller, S., Mancino, V., Offermanns, S., and Simon, M.I. (2002). Interaction of Gα12 with Gα13 and Gαq signaling pathways. Proc. Natl. Acad. Sci. USA 99, 9352–9357. Abstract Article
Hajdu-Cronin, Y.M., Chen, W.J., Patikoglou, G., Koelle, M.R., and Sternberg, P.W. (1999). Antagonism between Goα and Gqα in Caenorhabditis elegans: the RGS protein EAT-16 is necessary for Goα signaling and regulates Gqα activity. Genes Dev. 13, 1780–1793. Abstract
Hess, H.A., Roper, J.C., Grill, S.W., and Koelle, M.R. (2004). RGS-7 completes a receptor-independent heterotrimeric G protein cycle to asymmetrically regulate mitotic spindle positioning in C. elegans. Cell 119, 209–218. Abstract Article
Hilliard, M.A., Apicella, A.J., Kerr, R., Suzuki, H., Bazzicalupo, P., and Schafer, W.R. (2005). In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. EMBO J. 24, 63–72. Abstract Article
Hilliard, M.A., Bergamasco, C., Arbucci, S., Plasterk, R.H., and Bazzicalupo, P. (2004). Worms taste bitter: ASH neurons, QUI-1, GPA-3 and ODR-3 mediate quinine avoidance in Caenorhabditis elegans. EMBO J. 23, 1101–1111. Abstract Article
Hobson, R.J., Geng, J., Gray, A.D., and Komuniecki, R.W. (2003). SER-7b, a constitutively active Gαs coupled 5-HT7-like receptor expressed in the Caenorhabditis elegans M4 pharyngeal motorneuron. J. Neurochem. 87, 22–29 Abstract Article
Jansen, G., Thijssen, K.L., Werner, P., van der Horst, M., Hazendonk, E., and Plasterk, R.H. (1999). The complete family of genes encoding G proteins of Caenorhabditis elegans. Nat. Genet. 21, 414–419. Abstract Article
Jansen, G., Weinkove, D., and Plasterk, R.H. (2002). The G protein γ-subunit gpc-1 of the nematode C. elegans is involved in taste adaptation. EMBO J. 21, 986–994. Abstract Article
Jiang, H., Wu, D., and Simon, M.I. (1994). Activation of phospholipase Cβ4 by heterotrimeric GTP-binding proteins. J. Biol. Chem. 269, 7593–7596. Abstract
Jiang, L.I., and Sternberg, P.W. (1999). An HMG1-like protein facilitates Wnt signaling in Caenorhabditis elegans. Genes Dev. 13, 877–889. Abstract
Kamath, R.S., Fraser, A.G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. Abstract Article
Katoh, H., Aoki, J., Yamaguchi, Y., Kitano, Y., Ichikawa, A., and Negishi, M. (1998). Constitutively active Gα12, Gα13, and Gαq induce Rho-dependent neurite retraction through different signaling pathways. J. Biol. Chem. 273, 28700–28707. Abstract Article
Keane, J., and Avery, L. (2003). Mechanosensory inputs influence Caenorhabditis elegans pharyngeal activity via ivermectin sensitivity genes. Genetics 164, 153–162. Abstract
Kindt, K.S., Tam, T., Whiteman, S., and Schafer, W.R. (2002). Serotonin promotes Go-dependent neuronal migration in Caenorhabditis elegans. Curr. Biol. 12, 1738–1747. Abstract Article
Koelle, M.R., and Horvitz, H.R. (1996). EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 84, 115–125. Abstract Article
Kokel, M., Borland, C.Z., DeLong, L., Horvitz, H.R., and Stern, M.J. (1998). clr-1 encodes a receptor tyrosine phosphatase that negatively regulates an FGF receptor signaling pathway in Caenorhabditis elegans. Genes Dev. 12, 1425–1437. Abstract
Korswagen, H.C., Park, J.H., Ohshima, Y., and Plasterk, R.H. (1997). An activating mutation in a Caenorhabditis elegans Gs protein induces neural degeneration. Genes Dev. 11, 1493–1503. Abstract
Korswagen, H.C., van der Linden, A.M., and Plasterk, R.H. (1998). G protein hyperactivation of the Caenorhabditis elegans adenylyl cyclase SGS-1 induces neuronal degeneration. EMBO J. 17, 5059–5065. Abstract Article
Kozasa, T., Jiang, X., Hart, M.J., Sternweis, P.M., Singer, W.D., Gilman, A.G., Bollag, G., and Sternweis, P.C. (1998). p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13 [see comments]. Science 280, 2109–2111. Abstract Article
Lackner, M.R., Nurrish, S.J., and Kaplan, J.M. (1999). Facilitation of synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding to UNC-13 is required to stimulate acetylcholine release [see comments]. Neuron 24, 335–346. Abstract Article
Lans, H., Rademakers, S., and Jansen, G. (2004). A network of stimulatory and inhibitory Gα-subunits regulates olfaction in Caenorhabditis elegans. Genetics 167, 1677–1687. Abstract Article
Lochrie, M.A., Mendel, J.E., Sternberg, P.W., and Simon, M.I. (1991). Homologous and unique G protein α subunits in the nematode Caenorhabditis elegans. Cell Regul. 2, 135–154. Abstract
Lowry, W.E., Huang, J., Ma, Y.C., Ali, S., Wang, D., Williams, D.M., Okada, M., Cole, P.A., and Huang, X.Y. (2002). Csk, a critical link of G protein signals to actin cytoskeletal reorganization. Dev. Cell 2, 733–744. Abstract Article
Maeda, I., Kohara, Y., Yamamoto, M., and Sugimoto, A. (2001). Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol. 11, 171–176. Abstract Article
Manning, D.R. (2003). Evidence mounts for receptor-independent activation of heterotrimeric G proteins normally in vivo: positioning of the mitotic spindle in C. elegans. Sci. STKE 2003, pe35. Abstract Article
Mendel, J.E., Korswagen, H.C., Liu, K.S., Hajdu-Cronin, Y.M., Simon, M.I., Plasterk, R.H., and Sternberg, P.W. (1995). Participation of the protein Go in multiple aspects of behavior in C. elegans [see comments]. Science 267, 1652–1655. Abstract
Miller, K.G., Alfonso, A., Nguyen, M., Crowell, J.A., Johnson, C.D., and Rand, J.B. (1996). A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. USA 93, 12593–12598. Abstract Article
Miller, K.G., Emerson, M.D., and Rand, J.B. (1999). Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron 24, 323–333. Abstract Article
Miller, K.G., and Rand, J.B. (2000). A role for RIC-8 (Synembryn) and GOA-1 (Goα) in regulating a subset of centrosome movements during early embryogenesis in Caenorhabditis elegans. Genetics 156, 1649–1660. Abstract
Moghal, N., Garcia, L.R., Khan, L.A., Iwasaki, K., and Sternberg, P.W. (2003). Modulation of EGF receptor-mediated vulva development by the heterotrimeric G protein Gαq and excitable cells in C. elegans. Development 130, 4553–4566. Abstract Article
Moorman, C., and Plasterk, R.H. (2002). Functional characterization of the adenylyl cyclase gene sgs-1 by analysis of a mutational spectrum in Caenorhabditis elegans. Genetics 161, 133–142. Abstract
Nelson, L.S., Rosoff, M.L., and Li, C. (1998). Disruption of a neuropeptide gene, flp-1, causes multiple behavioral defects in Caenorhabditis elegans. Science 281, 1686–1690. Abstract Article
Neves, S.R., Ram, P.T., and Iyengar, R. (2002). G protein pathways. Science 296, 1636–1639. Abstract Article
Nurrish, S., Ségalat, L., and Kaplan, J.M. (1999). Serotonin inhibition of synaptic transmission: Gαo decreases the abundance of UNC-13 at release sites. Neuron 24, 231–242. Abstract Article
Park, J.H., Ohshima, S., Tani, T., and Ohshima, Y. (1997). Structure and expression of the gsa-1 gene encoding a G protein alpha αs subunit in C. elegans. Gene 194, 183–190. Abstract Article
Ranganathan, R., Sawin, E.R., Trent, C., and Horvitz, H.R. (2001). Mutations in the Caenorhabditis elegans serotonin reuptake transporter MOD-5 reveal serotonin-dependent and -independent activities of fluoxetine. J. Neurosci. 21, 5871–5884. Abstract
Reynolds, N.K., Schade, M.A., and Miller, K. (2005). Convergent, RIC-8 Dependent Gα Signaling Pathways in the C. elegans Synaptic Signaling Network. Genetics 169, 651–670. Abstract Article
Richmond, J.E., Davis, W.S., and Jorgensen, E.M. (1999). UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci. 2, 959–964. Abstract Article
Richmond, J.E., Weimer, R.M., and Jorgensen, E.M. (2001). An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412, 338–341. Abstract Article
Roayaie, K., Crump, J.G., Sagasti, A., and Bargmann, C.I. (1998). The Gα protein ODR-3 mediates olfactory and nociceptive function and controls cilium morphogenesis in C. elegans olfactory neurons. Neuron 20, 55–67. Abstract Article
Robatzek, M., Niacaris, T., Steger, K., Avery, L., and Thomas, J.H. (2001). eat-11 encodes GPB-2, a Gβ5 ortholog that interacts with Goα and Gqα to regulate C. elegans behavior. Curr. Biol. 11, 288–293. Abstract Article
Robatzek, M., and Thomas, J.H. (2000). Calcium/calmodulin-dependent protein kinase II regulates Caenorhabditis elegans locomotion in concert with a Go/Gq signaling network. Genetics 156, 1069–1082. Abstract
Rogers, C.M., Franks, C.J., Walker, R.J., Burke, J.F., and Holden-Dye, L. (2001). Regulation of the pharynx of Caenorhabditis elegans by 5-HT, octopamine, and FMRFamide-like neuropeptides. J. Neurobiol. 49, 235–244. Abstract Article
Sambongi, Y., Nagae, T., Liu, Y., Yoshimizu, T., Takeda, K., Wada, Y., and Futai, M. (1999). Sensing of cadmium and copper ions by externally exposed ADL, ASE, and ASH neurons elicits avoidance response in Caenorhabditis elegans. Neuroreport 10, 753–757. Abstract
Sawin, E.R., Ranganathan, R., and Horvitz, H.R. (2000). C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619–631. Abstract Article
Schade, M.A., Reynolds, N.K., Dollins, C.M., and Miller, K.G. (2005). Mutations that Rescue the Paralysis of C. elegans ric-8 (Synembryn) Mutants Activate the Gαs Pathway and Define a Third Major Branch of the Synaptic Signaling Network. Genetics 169, 631–649. Abstract Article
Ségalat, L., Elkes, D.A., and Kaplan, J.M. (1995). Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans [see comments]. Science 267, 1648–1651. Abstract
Shyn, S.I., Kerr, R., and Schafer, W.R. (2003). Serotonin and Go modulate functional states of neurons and muscles controlling C. elegans egg-laying behavior. Curr. Biol. 13, 1910–1915. Abstract Article
Simmer, F., Moorman, C., Van Der Linden, A.M., Kuijk, E., Van Den Berghe, P.V., Kamath, R., Fraser, A.G., Ahringer, J., and Plasterk, R.H. (2003). Genome-Wide RNAi of C. elegans Using the Hypersensitive rrf-3 Strain Reveals Novel Gene Functions. PLoS Biol. 1, E12. Abstract Article
Snow, B.E., Krumins, A.M., Brothers, G.M., Lee, S.F., Wall, M.A., Chung, S., Mangion, J., Arya, S., Gilman, A.G., and Siderovski, D.P. (1998). A G protein γ subunit-like domain shared between RGS11 and other RGS proteins specifies binding to Gβ5 subunits. Proc. Natl. Acad. Sci. USA 95, 13307–13312. Article
Srinivasan, D.G., Fisk, R.M., Xu, H., and van den Heuvel, S. (2003). A complex of LIN-5 and GPR proteins regulates G protein signaling and spindle function in C. elegans. Genes Dev. 17, 1225–1239. Abstract Article
Sternweis, P.C., and Smrcka, A.V. (1993). G proteins in signal transduction: the regulation of phospholipase C. Ciba Found Symp. 176, 96–106; discussion 106–111. Abstract
Sunahara, R.K., Dessauer, C.W., and Gilman, A.G. (1996). Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 36, 461–480. Abstract Article
Suzuki, N., Nakamura, S., Mano, H., and Kozasa, T. (2003). Gα12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl. Acad. Sci. U.S.A. 100, 733–738. Abstract Article
Syntichaki, P., Xu, K., Driscoll, M., and Tavernarakis, N. (2002). Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419, 939–944. Abstract Article
Tall, G.G., Krumins, A.M., and Gilman, A.G. (2003). Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362. Abstract Article
Trent, C., Tsung, N., and Horvitz, H.R. (1983). Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics 104, 619–647. Abstract
Tsou, M.F., Hayashi, A., and Rose, L.S. (2003). LET-99 opposes Gα /GPR signaling to generate asymmetry for spindle positioning in response to PAR and MES-1/SRC-1 signaling. Development 130, 5717–5730. Abstract Article
van der Linden, A.M., Moorman, C., Cuppen, E., Korswagen, H.C., and Plasterk, R.H. (2003). Hyperactivation of the G12-mediated signaling pathway in Caenorhabditis elegans induces a developmental growth arrest via protein kinase C. Curr. Biol. 13, 516–521. Abstract Article
van der Linden, A.M., Simmer, F., Cuppen, E., and Plasterk, R.H. (2001). The G protein β-subunit GPB-2 in Caenorhabditis elegans regulates the Goα -Gqα signaling network through interactions with the regulator of G protein signaling proteins EGL-10 and EAT-16. Genetics 158, 221–235. Abstract
van der Voorn, L., Gebbink, M., Plasterk, R.H., and Ploegh, H.L. (1990). Characterization of a G protein β-subunit gene from the nematode Caenorhabditis elegans. J. Mol. Biol. 213, 17–26. Abstract
van Swinderen, B., Metz, L.B., Shebester, L.D., and Crowder, C.M. (2002). A Caenorhabditis elegans pheromone antagonizes volatile anesthetic action through a Go-coupled pathway. Genetics 161, 109–119. Abstract
van Swinderen, B., Metz, L.B., Shebester, L.D., Mendel, J.E., Sternberg, P.W., and Crowder, C.M. (2001). Goα regulates volatile anesthetic action in Caenorhabditis elegans. Genetics 158, 643–655. Abstract
Voyno-Yasenetskaya, T.A., Pace, A.M., and Bourne, H.R. (1994). Mutant α-subunits of G12 and G13 proteins induce neoplastic transformation of Rat-1 fibroblasts. Oncogene 9, 2559–2565. Abstract
Waggoner, L.E., Hardaker, L.A., Golik, S., and Schafer, W.R. (2000). Effect of a neuropeptide gene on behavioral states in Caenorhabditis elegans egg-laying. Genetics 154, 1181–1192. Abstract Article
Walsh, D.A., and Van Patten, S.M. (1994). Multiple pathway signal transduction by the cAMP-dependent protein kinase. Faseb J. 8, 1227–1236. Abstract
Wang, Q., and Wadsworth, W.G. (2002). The C domain of netrin UNC-6 silences calcium/calmodulin-dependent protein kinase- and diacylglycerol-dependent axon branching in Caenorhabditis elegans. J. Neurosci. 22, 2274–2282. Abstract
Watson, N., Linder, M.E., Druey, K.M., Kehrl, J.H., and Blumer, K.J. (1996). RGS family members: GTPase-activating proteins for heterotrimeric G-protein α-subunits. Nature 383, 172–175. Abstract Article
Wickman, K., and Clapham, D.E. (1995a). Ion channel regulation by G proteins. Physiol. Rev. 75, 865–885. Abstract
Wickman, K.D., and Clapham, D.E. (1995b). G protein regulation of ion channels. Curr. Opin. Neurobiol. 5, 278–285. Abstract Article
Willson, J., Amliwala, K., Davis, A., Cook, A., Cuttle, M.F., Kriek, N., Hopper, N.A., O'Connor, V., Harder, A., Walker, R.J., and Holden-Dye, L. (2004). Latrotoxin Receptor Signaling Engages the UNC-13-Dependent Vesicle-Priming Pathway in C. elegans. Curr. Biol. 14, 1374–1379. Abstract Article
Xu, K., Tavernarakis, N., and Driscoll, M. (2001). Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca2+ release from the endoplasmic reticulum. Neuron 31, 957–971. Abstract Article
Yau, D.M., Yokoyama, N., Goshima, Y., Siddiqui, Z.K., Siddiqui, S.S., and Kozasa, T. (2003). Identification and molecular characterization of the Gα12-Rho guanine nucleotide exchange factor pathway in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 100, 14748–14753. Abstract Article
Zwaal, R.R., Ahringer, J., van Luenen, H.G., Rushforth, A., Anderson, P., and Plasterk, R.H. (1996). G proteins are required for spatial orientation of early cell cleavages in C. elegans embryos. Cell 86, 619–629. Abstract Article
Zwaal, R.R., Mendel, J.E., Sternberg, P.W., and Plasterk, R.H. (1997). Two neuronal G proteins are involved in chemosensation of the Caenorhabditis elegans Dauer-inducing pheromone. Genetics 145, 715–727. Abstract
*Edited by Iva Greenwald. Last revised October 11, 2006. Published October 13, 2006. This chapter should be cited as: Bastiani, C. and Mendel, J. Heterotrimeric G proteins in C. elegans (October 13, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.75.1, http://www.wormbook.org.
Copyright: © 2006 Carol Bastiani and Jane Mendel. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. mendelj@caltech.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.