Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
A wide variety of bacterial pathogens, as well as several fungi, kill C. elegans or produce non-lethal disease symptoms. This allows the nematode to be used as a simple, tractable model host for infectious disease. Human pathogens that affect C. elegans include Gram-negative bacteria of genera Burkholderia, Pseudomonas, Salmonella, Serratia and Yersinia; Gram-positive bacteria Enterococcus, Staphylococcus and Streptococcus; and the fungus Cryptococcus neoformans. Microbes that are not pathogenic to mammals, such as the insect pathogen Bacillus thuringiensis and the nematode-specific Microbacterium nematophilum, are also studied with C. elegans. Many of the pathogens investigated colonize the C. elegans intestine, and pathology is usually quantified as decreased lifespan of the nematode. A few microbes adhere to the nematode cuticle, while others produce toxins that kill C. elegans without a requirement for whole, live pathogen cells to contact the worm. The rapid growth and short generation time of C. elegans permit extensive screens for mutant pathogens with diminished killing, and some of the factors identified in these screens have been shown to play roles in mammalian infections. Genetic screens for toxin-resistant C. elegans mutants have identified host pathways exploited by bacterial toxins.
Nematodes began feeding on microbes long before the emergence of vertebrates. Microbial mechanisms to fend off nematodes - and perhaps even turn the animals into the microbe's food - must be of equally ancient origin. It is likely, then, that many of the microbial processes at work in human infectious diseases are adaptations of pathways that first evolved as defenses against nematodes. To the extent that this is the case, C. elegans can provide information about these virulence mechanisms that can be difficult to obtain with traditional animal models.
To fully understand a pathogen, researchers ideally would screen the microbe's genome comprehensively to identify all virulence genes. When mice or other mammals are the experimental host, such screens are impractical and prohibitively expensive, inasmuch as they require many thousands of animals. With C. elegans, the requirement for animals is not limiting because they grow rapidly in a simple and inexpensive culture system: production of 10,000 animals is trivial. As a result, a number of screens have been performed in which thousands of pathogen mutants have been tested individually against worms.
Of course, C. elegans offers far more than mere fecundity. Three decades of research has provided an extensive collection of mutants and clones that are available as off-the-shelf reagents for analysis of the host side of pathogen-host interactions. De novo genetic analysis of the host is feasible, e.g. by screening C. elegans for mutants that are either resistant or hypersensitive to a pathogen. Screens for hypersensitive mutants have been especially productive in elucidating the C. elegans innate immune system, as described in Signaling in the immune response by Jonathan J. Ewbank.
For convenience, pathogenic effects on C. elegans can be divided into two broad categories. In one, the nematode becomes infected, and presence of whole, live microbes within the animal or attached to the cuticle is correlated with death or disease. In the other, secreted microbial products, i.e. toxins, are responsible for symptoms, and C. elegans need not be exposed to live organisms. These categories are not mutually exclusive: one well-studied pathogen, Pseudomonas aeruginosa, has a "slow killing" mode involving colonization of C. elegans as well as two toxin-mediated pathways. Furthermore, it is not trivial to discern the mode of killing, and often pathogenesis appears to be multifactoral.
Table 1 lists microbial species known to be pathogenic to C. elegans, most of which are also human pathogens. Two of the pathogens (Drechmeria coniospora and Cryptococcus neoformans) are fungi, and the remainder are bacteria. Some of the bacteria, e.g. Yersinia sp. and Salmonella sp., are known to invade host cells during mammalian infections, but invasion of C. elegans cells has not been reported. No viruses or non-fungal eukaryotic parasites have been shown to infect or kill C. elegans.
Following are brief descriptions of the pathogens and their effects on C. elegans. In this chapter, "virulence factor" means any pathogen molecule involved in infection, including those that directly affect the host as well as those that function in the pathogen's own physiology, metabolism, and regulation. "Effector" and "toxin" refer to the subset of virulence factors that act directly on the host organism. For the sake of brevity, references have been omitted for general background on the pathogens.
Table 1.
| Organism | Human pathogen? | C. elegans pathology | References |
|---|---|---|---|
| Aeromonas hydrophila | Yes | Diminished lifespana | Couillault and Ewbank, 2002 |
| Agrobacterium tumefaciens | Yesb | Diminished lifespan[a] | Couillault and Ewbank, 2002 |
| Bacillus thuringiensis | No | Pore-forming toxin | Griffitts et al., 2003; Griffitts et al., 2001; Marroquin et al., 2000 |
| Burkholderia cenocepacia | Yes | Gut infection; toxin | Coenye et al., 2001; Kothe et al., 2003 |
| Burkholderia pseudomallei | Yes | Diminished lifespan[a]; toxin | Gan et al., 2002; O'Quinn et al., 2001 |
| Cryptococcus neoformans | Yes | Gut infection; toxin | Mylonakis et al., 2002; Mylonakis et al., 2004 |
| Drechmeria coniospora | No | Whole body parasitized | Jannson, 1994 |
| Enterococcus faecaelis | Yes | Gut infection | Garsin et al., 2001; Kim et al., 2002; Sifri et al., 2002 |
| Erwinia carotovora | Noc | Diminished lifespan[a] | Couillault and Ewbank, 2002 |
| Erwinia chrysanthemi | No[c] | Diminished lifespan[a] | Couillault and Ewbank, 2002 |
| Escherichia coli | Yes | Gut infection | Garsin et al., 2001 |
| Microbacterium nematophilum | No | Rectal & posterior cuticle infection | Hodgkin et al., 2000; Nicholas and Hodgkin, 2004 |
| Photorhabdus luminescens | Yes[b] | Diminished lifespan[a] | Couillault and Ewbank, 2002 |
| Pseudomonas aeruginosa | Yes | Gut infection; multiple toxins | Darby et al., 1999; Gallagher and Manoil, 2001; Kim et al., 2002; Mahajan-Miklos et al., 1999; Tan et al., 1999; Tan et al., 1999; Wareham et al., 2005 |
| Salmonella enterica | Yes | Gut infection; germline cell death | Aballay and Ausubel, 2001; Aballay et al., 2000; Labrousse et al., 2000; Tenor et al., 2004 |
| Serratia marcescens | Yes | Gut infection; tissue degradation | Kurz et al., 2003; Mallo et al., 2002 |
| Shewanella frigidimarina | No[c] | Diminished lifespan[a] | Couillault and Ewbank, 2002 |
| Shewanella massilia | No[c] | Diminished lifespan[a] | Couillault and Ewbank, 2002 |
| Staphylococcus aureus | Yes | Gut infection | Bae et al., 2004; Begun et al., 2005; Garsin et al., 2001; Sifri et al., 2003 |
| Streptococcus agalactiae (Group B) | Yes | Toxin (hydrogen peroxide) | Bolm et al., 2004 |
| Streptococcus dysgalactiae | Yes | Toxin (hydrogen peroxide) | Bolm et al., 2004 |
| Streptococcus mitis (Viridans group) | Yes | Toxin (hydrogen peroxide) | Bolm et al., 2004 |
| Streptococcus oralis | Yes | Toxin (hydrogen peroxide) | Bolm et al., 2004 |
| Streptococcus pneumoniae | Yes | Toxin (hydrogen peroxide) | Jansen et al., 2002 |
| Streptococcus pyogenes (Group A) | Yes | Toxin (hydrogen peroxide) | Jansen et al., 2002 |
| Xenorhabdus nematophila | No | Biofilm on cuticle; toxin | Couillault and Ewbank, 2002 |
| Yersinia pestis | Yes | Biofilm on cuticle, feeding blocked | Darby et al., 2002 |
| Yersinia pseudotuberculosis | Yes | Biofilm on cuticle, feeding blocked | Darby et al., 2002 |
|
a"Diminished lifespan" indicates that the organism's killing mode has not been determined in more detail. bRare reports of human infection by this species. cRare reports of human infection by some species in this genus. |
|||
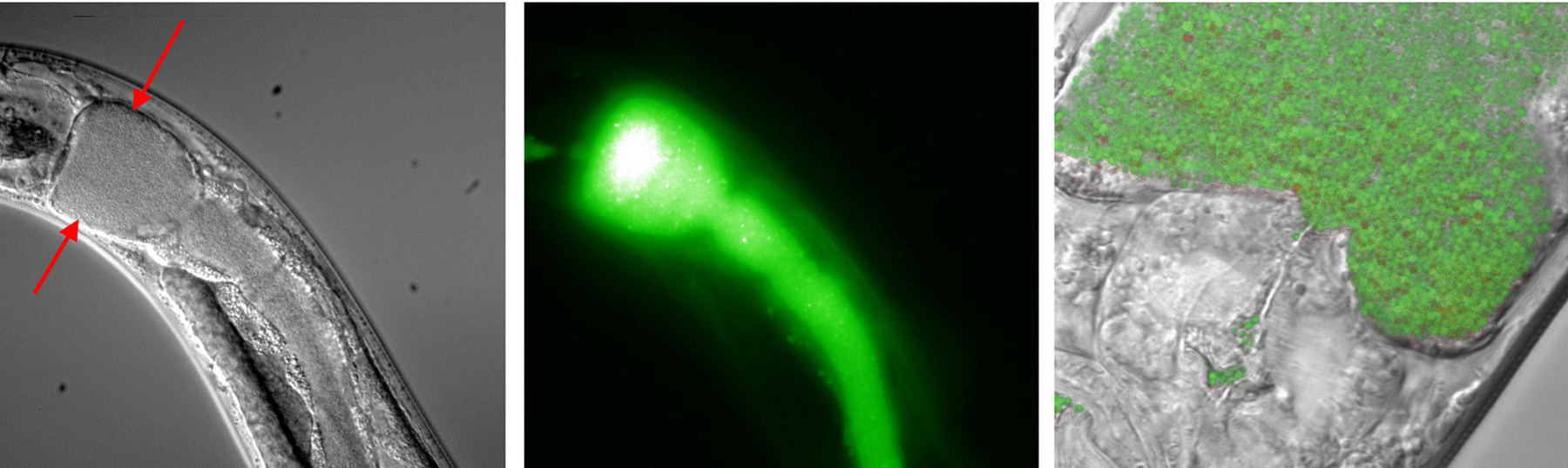
Numerous bacteria infect C. elegans. Experimenters take advantage of the fact that C. elegans eats microbes and simply place L4 or young adult animals on pathogen lawns to initiate infection. The transparency of C. elegans allows direct observation of infectious organisms within the worm by differential interference contrast microscopy (DIC); this is facilitated by using pathogens transformed to express green fluorescent protein (GFP; Figure 1). In many cases, the intestine becomes distended; it is not apparent whether this is due simply to physical pressure exerted by the growing pathogen or to some physiological response of the nematode. Colonization can be quantified by grinding worms into a homogenate, plating on solid media, and counting colonies. Mortality is usually reported as time for 50% of animals to die (LT50).
|
Figure 1. C. elegans colonized by Staphylococcus aureus expressing GFP. The anterior intestine is distended to the full width of the animal (arrows, left panel). Epifluorescence image of the same field (middle) shows that S. aureus colonizes the entire intestine. Individual bacteria can be resolved at high-magnification, as shown in merged bright-field and epifluorescence images (right). Photo: Jakob Begun and Frederick M. Ausubel.
E. coli OP50, the standard laboratory food, does not colonize wild-type C. elegans, but many pathogens do. Is intestinal colonization deleterious per se? It appears that the answer is no. Enterococcus faecium heavily colonizes the C. elegans gut, growing to a titer of more than 104 bacteria per nematode, without causing significant mortality (Garsin et al., 2001). This implies that when microbial killing occurs, there are specific pathogenic mechanisms at work.
E. faecalis is a Gram-positive organism that is part of the normal gut flora of many persons, but is also a serious pathogen that can cause meningitis, endocarditis and urinary tract infections. E. faecalis colonizes C. elegans and kills fairly rapidly, with an LT50 of about 4 days (d) (Garsin et al., 2001). Continuous feeding on E. faecalis is not necessary for heavy colonization; a small inoculum proliferates in the animal. Death was more rapid with E. faecalis expressing cytolysin, an effector that can lyse host cells. E. faecalis with a deletion in fsrB, part of a quorum-sensing system that responds to high culture density, was attenuated for killing, but colonized as efficiently as its wild-type parent.
Mutations in two protease genes under fsrB regulation, gelE and sprE, produced milder defects, suggesting that both fsrB-regulated effectors are involved in killing (Sifri et al., 2002). The fsrB mutant was also attenuated in a mouse model for peritonitis, establishing a connection between the C. elegans model and mammalian infection.
E. coli is known to C. elegans researchers primarily as worm food. The natural environment of E. coli is the digestive tract of warm-blooded animals, and in nature C. elegans is likely to encounter it only sporadically, e.g., in animal feces. In humans, E. coli can be either a harmless component of the natural gut flora or a serious pathogen.
C. elegans lived longer when grown on a common soil organism, Bacillus subtilis, than on the laboratory food strain of E. coli, OP50 (Garsin et al., 2003). Furthermore, when worms were fed OP50 grown on brain-heart infusion, a rich medium, their lifespan was shorter than with OP50 grown on standard NGM medium, a relatively poor source of bacterial nutrition (Garsin et al., 2001). These results suggest that E. coli is mildly pathogenic to C. elegans and that virulence increases on rich media. OP50 is a domesticated laboratory strain that lacks the O-antigen component of its outer membrane (S.L. Ananth and C.D., unpublished data), and is therefore not typical of pathogenic E. coli.
In mammalian infections, enteropathogenic E. coli (EPEC) produces attaching and effacing (AE) lesions in which microvilli of the small intestine are degraded and the bacteria intimately attach to the host epithelium. EPEC infection causes watery diarrhea in infants, which can lead to death by dehydration. C. elegans exposed to EPEC on NGM medium died prematurely, with an LT50 of 6.5 d, compared to 9.5 d for animals fed an E. coli K-12 control strain (J. Mellies, personal communication). After 24 h of exposure, EPEC titer was about 104 per nematode, versus about 102 for the controls. Microcolonies of EPEC expressing GFP were found primarily in the posterior intestine, and colonization was dependent on ler, a regulator required for AE lesions. However, C. elegans colonization does not require a type-three secretion system that injects bacterial proteins into host cytoplasm and is necessary for mammalian AE lesions. Bundle-forming pili, which adhere to mammalian host cells, also were not required in the worm. Furthermore, one EPEC clinical isolate killed C. elegans without colonizing, suggesting a toxin-mediated lethality.
Pseudomonas aeruginosa is a common soil and water bacterium that is pathogenic to both animals and plants. It rarely affects healthy persons, but can cause devastating infections in patients compromised by inborn genetic defects, acquired diseases, or injuries such as burns. P. aeruginosa lung infections are a major cause of death in patients with the genetic disease cystic fibrosis.
Depending on the strain and culture conditions, P. aeruginosa can kill C. elegans rapidly by toxin-mediated mechanisms (see below) or slowly in an infectious process. In the "slow killing" model, bacteria colonize the intestine, but during the first 24 h of exposure no strong disease symptoms are observed and mortality is negligible (Tan et al., 1999). With continued exposure, the worms gradually cease pharyngeal pumping, become immobile, then die. L4 larvae are more susceptible than adults, with LT50 of 38 h and 48 h, respectively. Direct observation using P. aeruginosa expressing GFP showed that bacteria began accumulating 36 h after feeding on the pathogen commenced. The lethal outcome could be prevented by removing the animals from P. aeruginosa after 12 h of exposure; longer periods before removal resulted in progressively poorer survival. Heat killed bacteria caused no mortality.
To identify bacterial factors involved in C. elegans killing, Tan et al. screened 2,400 transposon insertions in P. aeruginosa PA14 and recovered eight mutants (Tan et al., 1999). Four were severely attenuated, with LT50 greater than 90 h, and each contained an insertion in a regulatory gene. Two of these genes encode transcriptional activators, lasR and ptsP; the others, gacA and lemA, function together in a two-component signal transduction system. The requirement for LasR, the transcriptional activator of a "quorum sensing" system that responds to high bacterial cell density, is consistent with the observation that at least 12 h of colonization was required to produce a lethal infection. The factors under LasR control involved in slow killing have not been determined.
The eight mutations reducing virulence against C. elegans were tested in a mouse burn model under conditions that kill 100% of mice infected with wild-type P. aeruginosa (Tan et al., 1999). One mutant failed to kill any mice, another killed 100%, and the remaining six had intermediate levels of killing. Among the genes with reduced mouse killing was one which had no matches to known genes in databases at the time it was isolated. This suggests that the C. elegans slow killing model will be useful in identifying additional P. aeruginosa genes that are important in human infections.
Slow killing is likely to be multifactoral. A mutation eliminating exotoxin A, an enzyme that kills eukaryotic cells, was only moderately attenuated (LT50 55-70 h), suggesting that other effectors contribute to pathogenesis (Tan et al., 1999). Four effectors that in other systems attack host cytoplasmic functions (exoenzymes S, T, U and Y) are not required for slow killing (Wareham et al., 2005).
Salmonella is a genus of Gram-negative enteric bacteria that cause disease in a variety of vertebrates. In humans, S. enterica serovar Typhi causes the serious systemic infection known as typhoid fever; however, this organism is not highly pathogenic to mice. Conversely, S. enterica serovar Typhimurium is lethal to mice, causing a typhoid-like disease, but in humans causes nonfatal infection limited to the gastrointestinal tract. S. enterica serovar Typhimurium therefore has long been studied in the mouse as a model of systemic infection.
S. enterica serovar Typhimurium colonizes the C. elegans intestine (Aballay et al., 2000; Labrousse et al., 2000). For adults transferred to S. enterica and incubated at 25° C, the TD50 was 5.1 d, compared to 9.9 d for control animals on E. coli OP50 (Aballay et al., 2000). When worms were exposed to S. enterica for only 3 h, then removed to OP50, there was significant early death. Invasion of host cells is an essential aspect of Salmonella sp. pathogenesis in mammalian systems, but S. enterica does not appear to invade C. elegans cells.
S. enterica infection is accompanied by an increase in programmed cell death (PCD) in the germline (Aballay and Ausubel, 2001). This requires ced-3 and ced-4, indicating that the germline death involves the canonical PCD pathway that has been studied extensively in somatic cells (see Programmed cell death). However, there was no evidence that S. enterica induced somatic cell death. ced-3 and ced-4 mutants are more sensitive to S. enterica infection, suggesting that some PCD-related process is protective. Because the bacterial infection is limited to the intestine, it is not obvious how germline cell death itself could provide this protection. A pleiotropic S. enterica mutant, which lacks the phoP/phoQ virulence signaling pathway, failed to elicit germline cell death (Aballay and Ausubel, 2001), as did mutants for effector secretion (invH) and a transcriptional regulator (hilD; Tenor et al., 2004). Two other mutants, which are defective in lipopolysaccharide synthesis, exhibited reduced levels of killing and failed to trigger PCD, despite accumulating in the intestinal lumen (Aballay et al., 2003). In mammalian cell-culture systems, S. enterica SipB has been shown to be sufficient for apoptosis of host cells (Hersh et al., 1999); it has not been reported whether SipB is required for C. elegans germline cell death.
A screen of 960 transposon insertions in S. enterica produced 15 mutations with reduced killing of C. elegans, only some of which were in known virulence genes (Tenor et al., 2004). Some of the mutants were defective in assays for inducing neutrophil migration and invading epithelial cells, suggesting roles in mammalian pathogenesis. The genes represent a wide range of known or putative functions, including transcriptional activation, secretion, adhesion, outer-membrane synthesis and metabolism.
Serratia marcescens is a broad-host-range, opportunistic pathogen. This Gram-negative organism colonizes the C. elegans intestine, with an LT50 of approximately 4 d and complete killing in 6-7 d (Kurz et al., 2003). Early larvae are resistant to infection, but L4 larvae and adults are highly susceptible. S. marcescens infection of L4s increases the ability of E. coli cells to pass through the pharynx unlysed, suggesting that the pathogen disrupts the pharyngeal grinder. The infection also causes some intestinal cells to lyse and autofluorescent vacuoles to appear in the lumen. Neither heat-killed S. marcescens nor bacterial culture supernatants caused death, suggesting that killing requires live infection and that a diffusible toxin is not sufficient.
Kurz et al. (2003) screened for attenuated bacteria, identifying 19 loci out of 2,300 transposon insertions tested. Fewer than half of the mutants were attenuated in a Drosophila melanogaster model for S. marcescens, and only three were defective in a cytotoxicity assay against human epithelial cells. When the three were tested in a mouse lung infection model, only one was defective, and the mutated gene encodes a hemolysin that has been previously characterized.
Staphylococcus aureus causes a wide variety of diseases, including skin infections, food poisoning, toxic shock syndrome and septic shock. S. aureus kills all developmental stages of C. elegans (Sifri et al., 2003). Several strains have an LT50 of only about 1 d, making S. aureus among the swiftest of killers that infect the worm. Exposure of at least 8 h to the pathogen was required for killing.
Conflicting results were obtained by different laboratories regarding two well-studied S. aureus virulence regulators. Sifri et al. (2003) found strong attenuation with mutation of the agr locus and slight attenuation from a mutation in sarA. Working with a different S. aureus strain, Bae et al. (2004) reported that transposon insertions in either locus did not significantly reduce C. elegans killing. Both studies found that α-hemolysin, a secreted protein that lyses mammalian host cells, is important in C. elegans pathogenesis.
One group that screened for virulence mutations found 10 that were attenuated in more than one S. aureus strain background (Begun et al., 2005). Five of the genes identified had been found previously in mammalian models. The other five mutants were tested in a mouse model, and one was significantly attenuated. Because the transposon used for mutagenesis had a strong bias for one chromosomal region, the screen was not comprehensive.
Another group tested about 1,700 S. aureus transposon mutants and identified 71 genes, of which 41 could be assigned functions in metabolism, regulation, or production of extracellular factors (Bae et al., 2004). Only two extracellular factors, α-hemolysin and a secreted nuclease, were required for C. elegans killing. There was no attenuation in C. elegans killing for a strain defective in displaying about 20 surface proteins used in adhesion to mammalian hosts.
S. epidermidis is a Gram-positive pathogen that frequently grows as a biofilm, a population of microbes that adheres to a surface by means of an extracellular matrix the microbes secrete. S. epidermidis biofilms can grow on the surfaces of medical devices such as catheters and synthetic heart valves, sometimes leading to lethal infections. S. epidermidis infects and kills C. elegans, and killing requires the ica genes that code for synthesis of an extracellular polysaccharide biofilm component (J. Begun, C. Sifri and F. Ausubel, personal communication).
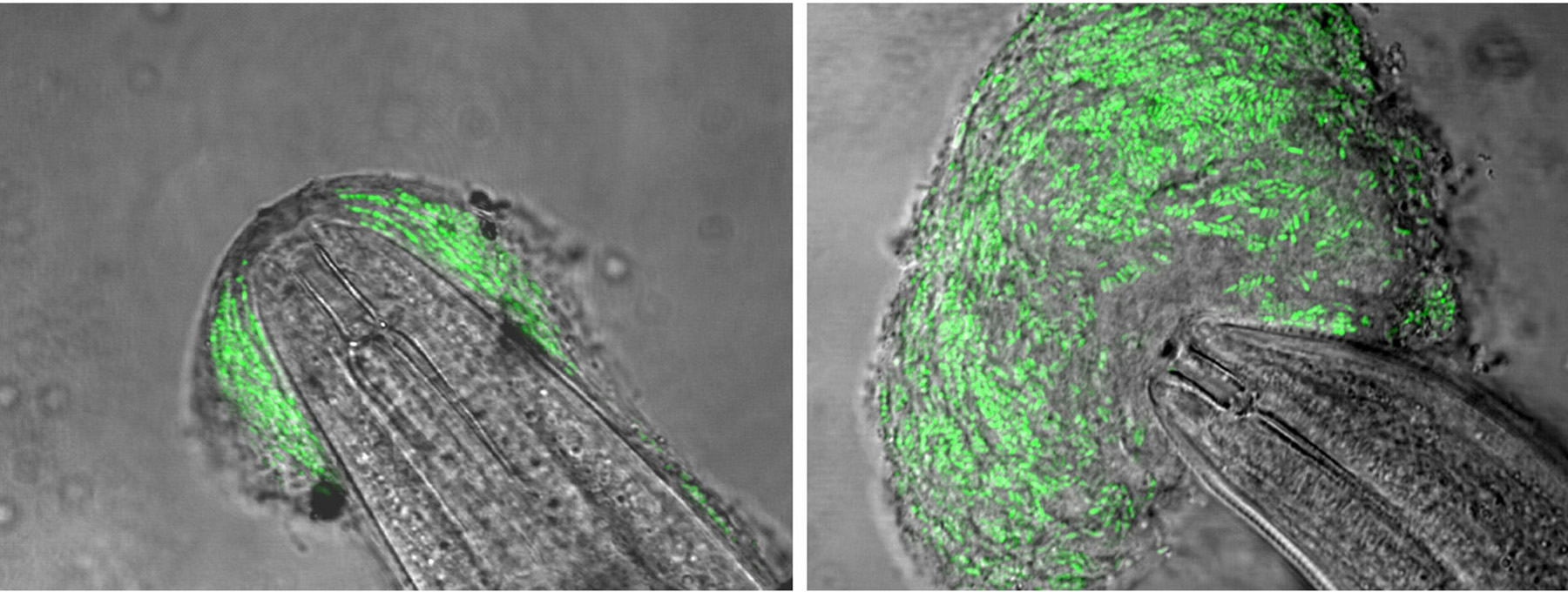
Microbacterium nematophilum is a Gram-positive organism whose pathogenic effect on C. elegans was discovered when the bacteria contaminated a laboratory culture (Hodgkin et al., 2000). Unlike most of the organism studied with C. elegans models, M. nematophilum is not known to be a pathogen of mammals.
M. nematophilum colonizes the C. elegans rectum, which is cuticle-lined, and a small peri-anal region of the exterior cuticle (Figure 2). The area around the anus becomes distended and swollen, a phenotype known as Dar (deformed anal region). The infection is not lethal to wild-type C. elegans, and the animals can grow and reproduce on a pure culture of these bacteria, although more slowly than on E. coli (Hodgkin et al., 2000). However, larvae of several other Caenorhabditis sp., including C. plicata and C. drosophilae, are highly susceptible and die within 24 h of exposure (J. Hodgkin, personal communication). Genetic methods have not been reported for M. nematophilum, and the mechanisms by which it induces Dar are unknown.
Because M. nematophilum attaches to the cuticle, mutants with altered surface properties were examined. srf-2, srf-3 and srf-5 animals were not colonized on the peri-anal cuticle and were not Dar (Hodgkin et al., 2000). Absence of Dar is known as Bus (bacterially unswollen).
The Dar response requires components of the ERK MAP kinase cascade (Nicholas and Hodgkin, 2004), which is also involved in cell fate specification. Infection without anal swelling is observed for worms with mutations encoding ERK pathway kinases (lin-45, mek-2, mpk-1), a scaffolding protein (ksr-1), and transcriptional activators (sur-2, lin-25). When infected, these mutants become severely constipated and their fertility is sharply reduced; this suggests that Dar is a defensive response of the nematode. Mutations in let-60, which activates the ERK pathway in development, did not produce Bus, suggesting a different means of activating ERK in response to M. nematophilum.
Screens for Bus mutants have identified 20 loci, and the mutants include alleles of sur-2 (Nicholas and Hodgkin, 2004) and srf-3 (Hoflich et al., 2004).
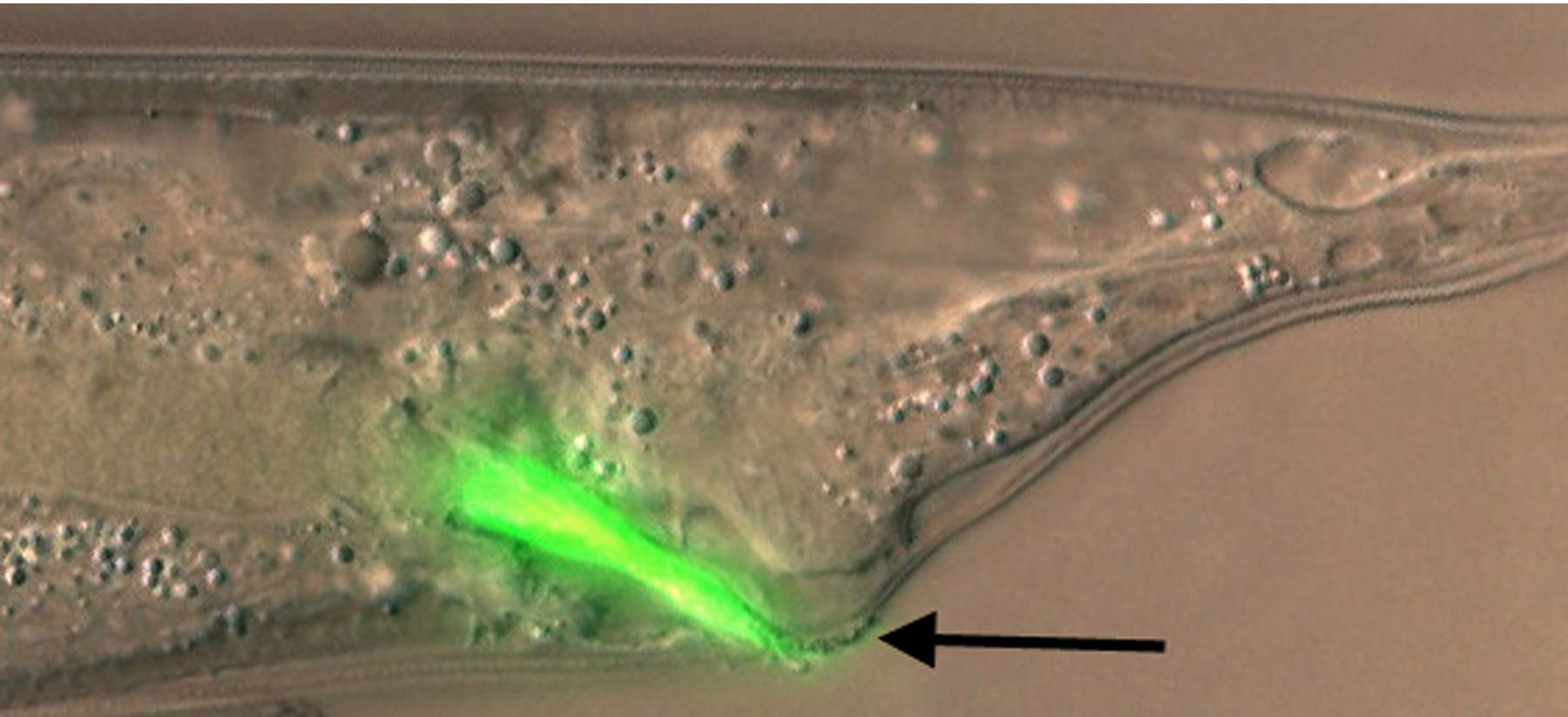
Y. pestis, the etiological agent of bubonic plague, and Y. pseudotuberculosis, a closely related pathogen that causes less serious infections, produce biofilms that attach to the cuticle of C. elegans, particularly on the head (Darby et al., 2002; Joshua et al., 2003). The biofilm matrix, which is polysaccharide rich (Tan and Darby, 2004), covers the mouth and blocks feeding (Figure 3), thereby inhibiting growth. This is similar to the manner in which Y. pestis colonizes and blocks feeding of its vector, the flea, to promote its transmission to mammals. Nematode biofilms and flea blockage both require Y. pestis hms genes that are involved in polysaccharide synthesis (Darby et al., 2002).
|
Figure 3. Biofilms on head formed by Yersinia pseudotuberculosis after one hour (left) or overnight (right) incubation on bacterial lawns. Nematodes were washed in M9 buffer before mounting for confocal epifluorescence microscopy. Yersinia cells express GFP; non-fluorescent material adhering to the head is bacterially-synthesized extracellular polysaccharide. Note that polysaccharide covers the mouth at one hour. Photo: Creg Darby.
C. elegans mutations in srf-2, srf-3 and srf-5, which change the surface characteristics of the worm, prevent biofilm attachment (Joshua et al., 2003), a phenotype known as Bah (biofilm absent on head). In a de novo screen for Bah mutants, an allele of srf-3 was obtained (Hoflich et al., 2004), as were alleles of srf-2, three bus genes, and three apparently novel genes, bah-1, bah-2 and bah-3 (author's unpublished results).
This section describes pathogen models in which the mode of killing, i.e. by infection or intoxication or both, has not been definitively determined, or in which both modes appear to be functional.
Burkholderia cepacia complex (BCC) is a group of closely related bacteria, including several that can infect the lungs of cystic fibrosis patients. BCC is taxonomically complex, with many ambiguities and changes in nomenclature (Coenye et al., 2001). In the two studies described in this subsection, different species names were used for the same strain; it is referred to here by the most recently used name, B. cenocepacia.
B. cenocepacia kills C. elegans slowly when the bacteria and worms are grown on standard media. GFP labeling of the bacteria showed extensive colonization of the intestine in this slow-killing assay, and death occurred in 1-4 d (Kothe et al., 2003). On high-osmolarity medium, B. cenocepacia killed completely within 24 h, and there was no detectable colonization in this fast killing. Conditioned media containing B. cenocepacia diffusible products caused some killing under high-osmolarity conditions; curiously, some worms were immobile and appeared dead at 4 h, but had recovered by 24 h. The toxin has not been identified, but cyanide, one of several P. aeruginosa toxins, was not detected in B. cenocepacia cultures.
Both modes of killing require the B. cenocepacia cep quorum-sensing regulatory system (Kothe et al., 2003). Mutation of a gene under quorum-sensing control, aidA, rendered the bacteria incapable of colonization and slow killing, but did not significantly affect fast killing.
Melioidosis, an infection that can be fatal, is caused by B. pseudomallei, a soil organism endemic in Southeast Asia and northern Australia. B. pseudomallei exposure causes C. elegans pharyngeal pumping to slow or cease, followed by a general decline in locomotion and eventual death (O'Quinn et al., 2001). The most virulent strain assayed had an LT50 of about 10 h. No assay for nematode colonization was reported. Curiously, B. pseudomallei cells inactivated by ultraviolet irradiation did not kill nematodes, but bacteria treated with high doses of gamma rays retained killing. The speed of the killing and the lethality of gamma-irradiated cultures suggests the action of a cell-free toxin. However, culture supernatants did not kill, nor did conditioned solid media, which contained diffusible products from bacterial growth but not live bacteria.
Another study, though, found killing from B. pseudomallei-conditioned media, although at reduced levels from that seen with whole cells (Gan et al., 2002). The decrease could mean that killing is multifactoral and includes processes that require live bacteria. Alternatively, toxin may be sufficient for killing, but less is available in conditioned media than when C. elegans is incubated continuously with live bacteria. As with the previous study, colonization of nematodes was not described. In this study, heat-killed bacteria did not kill nematodes, suggesting that the toxin is a protein or other heat labile substance.
Gan et al. screened 3,400 transposon insertion mutants of B. pseudomallei and obtained five with reduced killing of C. elegans. The five mutants were all attenuated to various degrees when inoculated intranasally into mice (Gan et al., 2002).
Couillault and Ewbank (2002) examined a variety of bacteria, including plant, fish and insect pathogens, assaying survival over time. Diminished lifespan resulted from exposure to a wide range, including plant pathogens Agrobacterium tumefaciens, Erwinia carotovora and Erwinia chrysanthemi; a marine bacterium that spoils stored fish, Shewanella frigidimarinia; and a freshwater fish pathogen that sometimes infects humans, Aeromonas hydrophila.
Photorhabdus luminescens and Xenorhabdus nematophila are related bacteria that live symbiotically in the intestines of Heterorhabditis sp. and Steinernema sp. nematodes, respectively. These entomopathogenic nematodes are parasites of insects, and it is their symbiotic bacteria that kill the insect host. Both Photorhabdus luminescens and Xenorhabdus nematophila were pathogenic to C. elegans (Couillault and Ewbank, 2002). Heat-killed X. nematophila retained significant killing ability, suggesting production of a heat-stable diffusible toxin. P. luminescens killing, while particularly rapid, was lost when bacteria were heat killed. X. nematophila colonized the head of C. elegans in a biofilm-like structure that resembles the one formed by Yersinia sp.
C. neoformans is a yeast-form fungus that can infect immunocompromised persons. When fed to C. elegans, the animals are colonized with a distended intestine and die with an LT50 of 5 - 7 d (Mylonakis et al., 2002). Two nonpathogenic cryptococci, C. laurentii and C. keutzingii, did not diminish the worm lifespan; in fact, animals lived longer on C. laurentii than on E. coli OP50. Several mutants known to be attenuated in mouse infections were also less pathogenic to C. elegans, and a mutation conferring hypervirulence in the mouse model killed C. elegans more rapidly than wild-type yeasts. These results suggest overlap between disease mechanisms in mammals and C. elegans.
Gut colonization is not essential for C. neoformans killing. Two C. neoformans strains that lack a polysaccharide capsule failed to accumulate in the intestine, but nevertheless killed C. elegans (Mylonakis et al., 2002). Heat-killed capsule-positive yeasts also killed, although more slowly than live fungi, but heat-killed capsule-negative strains were highly attenuated. These results suggest that a component of the C. neoformans capsule is toxic.
A screen of 350 C. neoformans insertion mutants yielded seven with attenuated virulence (Mylonakis et al., 2004). A mutation in kin1, encoding a serine/threonine kinase, did not affect C. elegans colonization but produced substantially less killing. The mutation did not alter previously studied C. neoformans virulence phenotypes, including capsule formation. When tested in mice by intranasal or intravenous injection, the kin1 mutant had a diminished ability to colonize several tissues, and killing was substantially slowed.
D. coniospora is an endoparasitic fungus that infects and consumes a narrow range of nematodes (Jannson, 1994). Conidia, which are spores, attach to the C. elegans head at the labial and amphid sensilla, and also attach to the vulva. The conidia produce threadlike hyphae that grow throughout the animal and consume it. When infection begins at the head, it is not apparent whether the fungus penetrates the cuticle or simply bypasses it by way of the sensilla openings. Protease treatment of worms before exposure decreased conidia attachment, suggesting involvement of nematode surface or secreted proteins. D. coniospora were able to bind che-12 and che-14 mutants, which have defective amphids; conidia also bound mec-1 animals, which have displaced amphid neurons, and vab-1 mutants, which have malformed heads.
Couillault et al. (2004) reported specific induction of antimicrobial peptide genes in C. elegans after infection by D. coniospora. One of these, nlp-31, encodes a peptide with demonstrable antifungal activity against D. coniospora.
Bacillus thuringiensis (Bt) is a Gram-positive bacterium used in insect control because it makes toxins that do not affect vertebrates. Bt expressing a single pore-forming toxin, Cry5B, causes intestinal damage to C. elegans in a manner that resembles the pathology observed in toxin-treated insects (Marroquin et al., 2000). The animals turn pale, the intestine becomes pitted, vacuolated and constricted, and brood size is reduced. There are no obvious effects on the neuromuscular systems of the worm. Expression of Cry5B in E. coli recapitulated these results, indicating that the damage is toxin-specific, not a general response to Bt.
Screening for C. elegans bre (Bacillus toxin resistant) mutants identified five genes (Marroquin et al., 2000). bre-2, bre-3, bre-4, bre-5 encode glycosyltransferases that appear to function in a single pathway and are required for intestinal cells to take up Cry5B (Griffitts et al., 2003; Griffitts et al., 2001). Binding of Cry5B to glycolipids that are absent in bre-3, bre-4 and bre-5 mutants has been demonstrated (Griffitts et al., 2005).
P. aeruginosa kills C. elegans in two distinct toxin-mediated modes. In the first, known as "fast killing," bacterial strain PA14 is used, and a high osmolarity medium is required (Mahajan-Miklos et al., 1999). At least 50%, and in some assays about 90%, of L4 animals died within 4 h of exposure. Killing is mediated by diffusible toxins and does not require contact with live bacteria.
A screen of 3,300 P. aeruginosa transposon insertions produced seven mutants with reduced killing (Mahajan-Miklos et al., 1999). Four mutants had reduced levels of pyocyanin, a pigmented secondary metabolite belonging to the phenazine family of tricyclic compounds. However, three killing-defective mutants made normal amounts of pyocyanin. Together, these results suggest that toxicity is multifactoral, involving pyocyanin (or other phenazines) and unidentified other molecules.
Pyocyanin is a redox-active compound toxic to eukaryotic cells, suggesting that fast killing is due at least in part to oxidative stress. Analysis of C. elegans mutants lent support to this hypothesis. age-1 mutants, which are resistant to other forms of oxidative stress, were significantly resistant to fast killing (Mahajan-Miklos et al., 1999). Conversely, mev-1 and rad-8 mutants, which are hypersensitive to oxidative stress, were more sensitive in fast-killing assays.
The C. elegans genes pgp-1 and pgp-3 encode P-glycoprotein membrane transporters. A pgp-1;pgp-3 double mutant was sensitive to fast killing without the need for high osmolarity (Mahajan-Miklos et al., 1999). Furthermore, the double mutant was hypersensitive to P. aeruginosa mutants that retained phenazine production, but was as resistant as wild type to phenazine-deficient mutants.
The second toxin-mediated killing model uses a different P. aeruginosa strain, PAO1. When PAO1 is grown to high density on an especially rich medium, animals placed on the lawn cease pharyngeal pumping almost instantaneously, become progressively paralyzed, and die within two hours (Darby et al., 1999). These effects are mediated by bacterially produced cyanide, a toxin that blocks respiratory electron transport (Gallagher and Manoil, 2001).
In a screen for C. elegans resistant to cyanide-mediated killing, two alleles of egl-9 were recovered (Darby et al., 1999). Although egl-9 worms were completely resistant to killing by live bacteria, dose-response experiments in vitro showed that the mutants can be killed by sufficiently high cyanide concentrations (Gallagher and Manoil, 2001). egl-9 encodes a dioxygenase that functions as a negative regulator of a hypoxia-response pathway (Epstein et al., 2001). This suggests that in egl-9 mutants, cyanide resistance is mediated by constitutive activation of the pathway. The role of cyanide in human P. aeruginosa infections, if any, is not known.
S. pneumoniae is a Gram-positive organism whose only natural reservoir is humans. Many persons are colonized asymptomatically, but the bacterium can also cause pneumonia and meningitis. S. pneumoniae kills C. elegans rapidly (Garsin et al., 2001), but does not colonize the worm, and bacterially synthesized hydrogen peroxide is sufficient for killing (Jansen et al., 2002). Other Streptococci that kill C. elegans with hydrogen peroxide include S. pyogenes (also known as group A Streptococcus) and S. agalactiae (group B; Bolm et al., 2004).
Pathogenic bacteria produce a wide variety of effectors that act on the host, and often the targets have conserved eukaryotic functions. Organisms that produce these effectors nevertheless may not be pathogenic to C. elegans because disease requires a variety of other functions, e.g. sensing of the host environment, gene induction, binding to host receptors, and evasion of host defenses. A strategy to overcome these limitations is to express bacterial effectors directly in transgenic C. elegans. This approach was demonstrated using pertussis toxin (PTX), a well-characterized effector known to enzymatically inactivate a class of heterotrimeric G proteins. Expression of PTX produced a phenotype indistinguishable from that of loss-of-function mutations in goa-1, which encodes the only PTX target in C. elegans (Darby and Falkow, 2001). PTX expression also suppressed a gain-of-function goa-1 mutation.
A mutation in the Salmonella enterica gene sptP was recovered in a screen for mutants with defects in killing C. elegans (Tenor et al., 2004). In mammals, SptP is injected into host cells, where it reverses cytoskeletal disruption produced earlier by another S. enterica effector (Fu and Galan, 1999). Transgenic animals were generated with sptP under the promoter of the intestinal gene mtl-2. The animals were then exposed to either S. enterica or Pseudomonas aeruginosainfection to determine whether ectopic expression of SptP enhances pathogenicity. In both cases a slight increase was observed in relative mortality (ratio of LT50 on pathogen to LT50 on E. coli) compared to untransformed controls.
The first paper describing C. elegans as a model host for a microbial pathogen was published in 1999. The field has grown rapidly since, and now investigates a diverse range of human pathogens that includes numerous bacteria and one fungus. Several microbes that are pathogenic to C. elegans but not to humans also are being examined.
As a rapidly growing, inexpensive host, C. elegans permits large-scale screens of pathogen genomes, and thousands of bacterial mutants have been tested in screens of P. aeruginosa (Gallagher and Manoil, 2001; Mahajan-Miklos et al., 1999; Tan et al., 1999), B. pseudomallei (Gan et al., 2002), S. marcescens (Kurz et al., 2003) and S. aureus (Bae et al., 2004). Some of the mutations recovered have been in virulence genes already known from mammalian studies, which validates the screening strategy (Bae et al., 2004; Kurz et al., 2003; Sifri et al., 2003; Tan et al., 1999; Tenor et al., 2004).
The ultimate goal is to discover new virulence factors for mammalian pathogenesis, and here results have been mixed. Some of the P. aeruginosa (Tan et al., 1999) and B. pseudomallei (Gan et al., 2002) mutants defective against C. elegans were also attenuated in a mouse models. On the other hand, no new mammalian virulence mutants were found in a screen of S. marcescens (Kurz et al., 2003), and investigators who screened S. aureus concluded that for this pathogen, there is limited overlap between C. elegans and mammalian pathogenesis (Bae et al., 2004).
With infection models, some screens for bacterial mutant appear biased toward identifying regulators. For instance, five of eight P. aeruginosa mutations in the slow killing model were in genes known or inferred to encode regulators (Tan et al., 1999). This suggests that slow killing is multifactoral, and that regulators are identified because mutations disrupt expression of several downstream factors. Screens in a multifactoral system may not have sufficient resolution to identify individual effectors. However, this drawback may be pathogen-specific, because a screen of S. enterica produced a mutation in sptP, which is known from mammalian studies to encode an effector that acts in the cytoplasm of host cells (Tenor et al., 2004).
In toxin-mediated killing models, screens of P. aeruginosa have proven useful in characterizing the toxins. A mutation in the bacterial hcnC gene, encoding hydrogen cyanide synthase, prompted experiments showing that cyanide is necessary and sufficient for lethal paralysis (Gallagher and Manoil, 2001). In the fast-killing model, mutations in phzB prompted further investigation of phenazines as candidate toxins (Mahajan-Miklos et al., 1999).
On the C. elegans side of the interaction, genetics can provide information about the host pathways that pathogens exploit. A notable success is the work of Aroian and colleagues on C. elegans mutants resistant to a Bt toxin. The screen identified a set of glycosyltransferases that function together in C. elegans (Griffitts et al., 2003; Griffitts et al., 2001; Marroquin et al., 2000), and this in turn facilitated identification of a glycolipid toxin receptor (Griffitts et al., 2005). However, screens to find resistant organisms do not always identify the pathways that interact directly with the pathogen. A screen for worms resistant to P. aeruginosa paralysis produced alleles of egl-9 (Darby et al., 1999); later characterization suggested that the resistance phenotype results from activation of a bypass pathway (Epstein et al., 2001).
Because C. elegans models for microbial pathogenesis are still new, the extent to which they are relevant to human disease remains to be determined. Obviously the overlap will not be complete, but this is true for any animal model. It is encouraging that some virulence gene discovered using C. elegans have been found to be required for full pathogenesis in mouse infections. These genes have not yet been characterized thoroughly, however. The task ahead is to take novel results from C. elegans systems and determine their importance in human infectious disease.
I thank Fred Ausubel, Jake Begun, Jonathan Hodgkin, Jay Mellies, Hannah Nicholas, Delia O'Rourke and Costi Sifri for communicating unpublished results. Kevin Drace, Michael A. Miller and two anonymous reviewers provided helpful comments on the manuscript. My laboratory is supported by National Institutes of Health grant AI057512.
Aballay, A., and Ausubel, F.M. (2001). Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc. Natl. Acad. Sci. USA 98, 2735–2739. Abstract Article
Aballay, A., Drenkard, E., Hilbun, L.R., and Ausubel, F.M. (2003). Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 13, 47–52. Abstract Article
Aballay, A., Yorgey, P., and Ausubel, F.M. (2000). Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr. Biol. 10, 1539–1542. Abstract Article
Bae, T., Banger, A.K., Wallace, A., Glass, E.M., Aslund, F., Schneewind, O., and Missiakas, D.M. (2004). Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. USA 101, 12312–12317. Abstract Article
Begun, J., Sifri, C.D., Goldman, S., Calderwood, S.B., and Ausubel, F.M. (2005). Staphylococcus aureus virulence factors identified by using a high-throughput Caenorhabditis elegans-killing model. Infect Immun. 73, 872–877. Abstract Article
Bolm, M., Jansen, W.T., Schnabel, R., and Chhatwal, G.S. (2004). Hydrogen peroxide-mediated killing of Caenorhabditis elegans: a common feature of different streptococcal species. Infect Immun. 72, 1192–1194. Abstract Article
Coenye, T., Vandamme, P., Govan, J.R., and LiPuma, J.J. (2001). Taxonomy and identification of the Burkholderia cepacia complex. J. Clin. Microbiol. 39, 3427–3436. Abstract Article
Couillault, C., and Ewbank, J.J. (2002). Diverse bacteria are pathogens of Caenorhabditis elegans. Infect. Immun. 70, 4705–4707. Abstract Article
Couillault, C., Pujol, N., Reboul, J., Sabatier, L., Guichou, J.F., Kohara, Y., and Ewbank, J.J. (2004). TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol. 5, 488–494. Abstract Article
Darby, C., Cosma, C.L., Thomas, J.H., and Manoil, C. (1999). Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 96, 15202–15207. Abstract Article
Darby, C., and Falkow, S. (2001). Mimicry of a G protein mutation by pertussis toxin expression in transgenic Caenorhabditis elegans. Infect Immun. 69, 6271–6275. Abstract Article
Darby, C., Hsu, J.W., Ghori, N., and Falkow, S. (2002). Caenorhabditis elegans: plague bacteria biofilm blocks food intake. Nature 417, 243–244. Abstract Article
Epstein, A.C., Gleadle, J.M., McNeill, L.A., Hewitson, K.S., O'Rourke, J., Mole, D.R., Mukherji, M., Metzen, E., Wilson, M.I., Dhanda, A., et al. (2001). C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54. Abstract Article
Fu, Y., and Galan, J.E. (1999). A Salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 401, 293–297. Abstract Article
Gallagher, L.A., and Manoil, C. (2001). Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J. Bacteriol. 183, 6207–6214. Abstract Article
Gan, Y.H., Chua, K.L., Chua, H.H., Liu, B., Hii, C.S., Chong, H.L., and Tan, P. (2002). Characterization of Burkholderia pseudomallei infection and identification of novel virulence factors using a Caenorhabditis elegans host system. Mol. Microbiol. 44, 1185–1197. Abstract Article
Garsin, D.A., Sifri, C.D., Mylonakis, E., Qin, X., Singh, K.V., Murray, B.E., Calderwood, S.B., and Ausubel, F.M. (2001). A simple model host for identifying Gram-positive virulence factors. Proc. Natl. Acad. Sci. USA 98, 10892–10897. Abstract Article
Garsin, D.A., Villanueva, J.M., Begun, J., Kim, D.H., Sifri, C.D., Calderwood, S.B., Ruvkun, G., and Ausubel, F.M. (2003). Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300, 1921. Abstract Article
Griffitts, J.S., Haslam, S.M., Yang, T., Garczynski, S.F., Mulloy, B., Morris, H., Cremer, P.S., Dell, A., Adang, M.J., and Aroian, R.V. (2005). Glycolipids as receptors for Bacillus thuringiensis crystal toxin. Science 307, 922–925. Abstract Article
Griffitts, J.S., Huffman, D.L., Whitacre, J.L., Barrows, B.D., Marroquin, L.D., Muller, R., Brown, J.R., Hennet, T., Esko, J.D., and Aroian, R.V. (2003). Resistance to a bacterial toxin is mediated by removal of a conserved glycosylation pathway required for toxin-host interactions. J. Biol. Chem. 278, 45594–45602. Abstract Article
Griffitts, J.S., Whitacre, J.L., Stevens, D.E., and Aroian, R.V. (2001). Bt toxin resistance from loss of a putative carbohydrate-modifying enzyme. Science 293, 860–864. Abstract Article
Hersh, D., Monack, D.M., Smith, M.R., Ghori, N., Falkow, S., and Zychlinsky, A. (1999). The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA 96, 2396–2401. Abstract Article
Hodgkin, J., Kuwabara, P.E., and Corneliussen, B. (2000). A novel bacterial pathogen, Microbacterium nematophilum, induces morphological change in the nematode C. elegans. Curr. Biol. 10, 1615–1618. Abstract Article
Hoflich, J., Berninsone, P., Gobel, C., Gravato-Nobre, M.J., Libby, B.J., Darby, C., Politz, S.M., Hodgkin, J., Hirschberg, C.B., and Baumeister, R. (2004). Loss of srf-3-encoded nucleotide sugar transporter activity in Caenorhabditis elegans alters surface antigenicity and prevents bacterial adherence. J. Biol. Chem. 279, 30440–30448. Abstract Article
Jannson, H.-B. (1994). Adhesion of conidia of Drechmeria coniospora to Caenorhabditis elegans wild type and mutants. J. Nematol. 26, 430–435.
Jansen, W.T., Bolm, M., Balling, R., Chhatwal, G.S., and Schnabel, R. (2002). Hydrogen peroxide-mediated killing of Caenorhabditis elegans by Streptococcus pyogenes. Infect Immun. 70, 5202–5207. Abstract Article
Joshua, G.W., Karlyshev, A.V., Smith, M.P., Isherwood, K.E., Titball, R.W., and Wren, B.W. (2003). A Caenorhabditis elegans model of Yersinia infection: biofilm formation on a biotic surface. Microbiology 149, 3221–3229. Abstract Article
Kim, D.H., Feinbaum, R., Alloing, G., Emerson, F.E., Garsin, D.A., Inoue, H., Tanaka-Hino, M., Hisamoto, N., Matsumoto, K., Tan, M.W., et al. (2002). A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623–626. Abstract Article
Kothe, M., Antl, M., Huber, B., Stoecker, K., Ebrecht, D., Steinmetz, I., and Eberl, L. (2003). Killing of Caenorhabditis elegans by Burkholderia cepacia is controlled by the cep quorum-sensing system. Cell Microbiol. 5, 343–351. Abstract Article
Kurz, C.L., Chauvet, S., Andres, E., Aurouze, M., Vallet, I., Michel, G.P., Uh, M., Celli, J., Filloux, A., De Bentzmann, S., et al. (2003). Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. EMBO J. 22, 1451–1460. Abstract Article
Labrousse, A., Chauvet, S., Couillault, C., Kurz, C.L., and Ewbank, J.J. (2000). Caenorhabditis elegans is a model host for Salmonella typhimurium. Curr. Biol. 10, 1543–1545. Abstract Article
Mahajan-Miklos, S., Tan, M.W., Rahme, L.G., and Ausubel, F.M. (1999). Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96, 47–56. Abstract Article
Mallo, G.V., Kurz, C.L., Couillault, C., Pujol, N., Granjeaud, S., Kohara, Y., and Ewbank, J.J. (2002). Inducible antibacterial defense system in C. elegans. Curr. Biol. 12, 1209–1214. Abstract Article
Marroquin, L.D., Elyassnia, D., Griffitts, J.S., Feitelson, J.S., and Aroian, R.V. (2000). Bacillus thuringiensis (Bt) toxin susceptibility and isolation of resistance mutants in the nematode Caenorhabditis elegans. Genetics 155, 1693–1699. Abstract
Mylonakis, E., Ausubel, F.M., Perfect, J.R., Heitman, J., and Calderwood, S.B. (2002). Killing of Caenorhabditis elegans by Cryptococcus neoformans as a model of yeast pathogenesis. Proc. Natl. Acad. Sci. USA 99, 15675–15680. Abstract Article
Mylonakis, E., Idnurm, A., Moreno, R., El Khoury, J., Rottman, J.B., Ausubel, F.M., Heitman, J., and Calderwood, S.B. (2004). Cryptococcus neoformans Kin1 protein kinase homologue, identified through a Caenorhabditis elegans screen, promotes virulence in mammals. Mol. Microbiol. 54, 407–419. Abstract Article
Nicholas, H.R., and Hodgkin, J. (2004). The ERK MAP kinase cascade mediates tail swelling and a protective response to rectal infection in C. elegans. Curr. Biol. 14, 1256–1261. Abstract Article
O'Quinn, A.L., Wiegand, E.M., and Jeddeloh, J.A. (2001). Burkholderia pseudomallei kills the nematode Caenorhabditis elegans using an endotoxin-mediated paralysis. Cell Microbiol. 3, 381–393. Abstract Article
Sifri, C.D., Begun, J., Ausubel, F.M., and Calderwood, S.B. (2003). Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect Immun. 71, 2208–2217. Abstract Article
Sifri, C.D., Mylonakis, E., Singh, K.V., Qin, X., Garsin, D.A., Murray, B.E., Ausubel, F.M., and Calderwood, S.B. (2002). Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice. Infect Immun. 70, 5647–5650. Abstract Article
Tan, L., and Darby, C. (2004). A movable surface: formation of Yersinia sp. biofilms on motile Caenorhabditis elegans. J. Bacteriol. 186, 5087–5092. Abstract Article
Tan, M.W., Mahajan-Miklos, S., and Ausubel, F.M. (1999). Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. USA 96, 715–720. Abstract Article
Tan, M.W., Rahme, L.G., Sternberg, J.A., Tompkins, R.G., and Ausubel, F.M. (1999). Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc. Natl. Acad. Sci. USA 96, 2408–2413. Abstract Article
*Edited by David H.A. Fitch. Last revised March 29, 2005. Published September 6, 2005. This chapter should be cited as: Darby, C. Interactions with microbial pathogens (September 6, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.21.1, http://www.wormbook.org.
Copyright: © 2005 Creg Darby. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: creg@uab.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.