Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergSydney Brenner originally conceived Caenorhabditis elegans as a model system for building a fundamental understanding of animal genetics, development and behavior, but it is also rapidly becoming a respectable model for evolutionary studies. Although C. elegans molecular biologists have long appreciated the value of sequence comparisons to identify conserved regions as indicators of function, arguably the most interesting aspects of evolution are changes in molecular functions, domains of expression, and developmental roles. Elucidating how such changes have shaped functional diversity at a variety of levels also has potential for augmenting our understanding of genome function and developmental mechanisms. Reciprocally, information from this model system facilitates studies of evolutionary pattern and process. The success of such comparative approaches to enhance our understanding obviously depends upon the availability of material and information from multiple related species, as well as different wild populations of C. elegans. This section of WormBook reviews much of the recent work to establish a foundation of evolutionary biology and ecology for C. elegans and other "satellites" of this model system.
Knowing the phylogenetic relationships between C. elegans and other nematodes or animals is important for comparative analyses of behavior, morphology, development, molecular mechanisms, and genomics. A phylogeny allows the reconstruction of the historical changes that have led to current variation and provides a way to test how often convergent changes have occurred. Species phylogenies are also crucial for bioinformatic analyses of genomes; they provide a basis for selecting species for comparative genomic sequencing and for testing orthologous and paralogous relationships in gene phylogenies, an important foundation for genome annotation and prediction of gene function (Eisen, 1998; Eisen and Fraser, 2003). A quick tour of nematode diversity and the backbone of nematode phylogeny provides a short tour of general nematode diversity and phylogeny. The phylogenetic relationships of C. elegans and other rhabditids reviews what is known so far about the closer relationships within the rhabditids and within genus Caenorhabditis in particular. The relationship of C. elegans to other animals or other major model systems, however, is not covered in these chapters, and is presented in a separate part of this introduction ("Animal Relationships" below).
There is substantial variation among rhabditids at genetic and developmental levels. Reconstructing how this variation arose is likely to illuminate how developmentally robust systems can nevertheless be modified by evolutionary change, one of the most intriguing and fundamental questions in evolutionary biology today. Nematode genome evolution reviews work on the evolution of genome organization and chromosome architecture. Evolution of development in nematodes related to C. elegans provides an overview of comparative developmental biology using C. elegans and other satellite model organisms, such as Pristionchus pacificus. These comparative studies reveal that genome architecture and developmental mechanisms can be strikingly diverse, suggesting that there is significant plasticity and potential variation available to selection, even for features that superficially appear to be highly conserved.
Despite remarkable progress toward a molecular understanding for C. elegans, an ecological context for this model system has lagged behind. Substantial molecular work has contributed to a detailed picture of the mechanisms involved in dauer formation, chemosensation and other aspects of neurobiology and behavior, innate immunity, germline development and sex determination, senescence and developmental timing. Although these features must somehow be important for the organism's life and its interaction with the environment, the actual ecological context and evolutionary "pressures" to which such features are adaptations have remained quite unclear. Natural variation and population genetics of C. elegans reviews work on C. elegans population genetics which will help us comprehend, among other things, how this organism migrates, the possible role of mating in its natural setting, how variation is related to geography and environment, and the birth-death cycles of populations. Ecology of Caenorhabditis species reviews the potential interactions that C. elegans may have with bacteria and pathogens in its environment. Interactions with microbial pathogens reviews current understanding of the ecological context of C. elegans and its closest relatives. Future work in these areas is likely to clarify why the biological features of C. elegans we like to study in the lab actually exist and why they may be important for the animal itself, thus providing further functional insight.
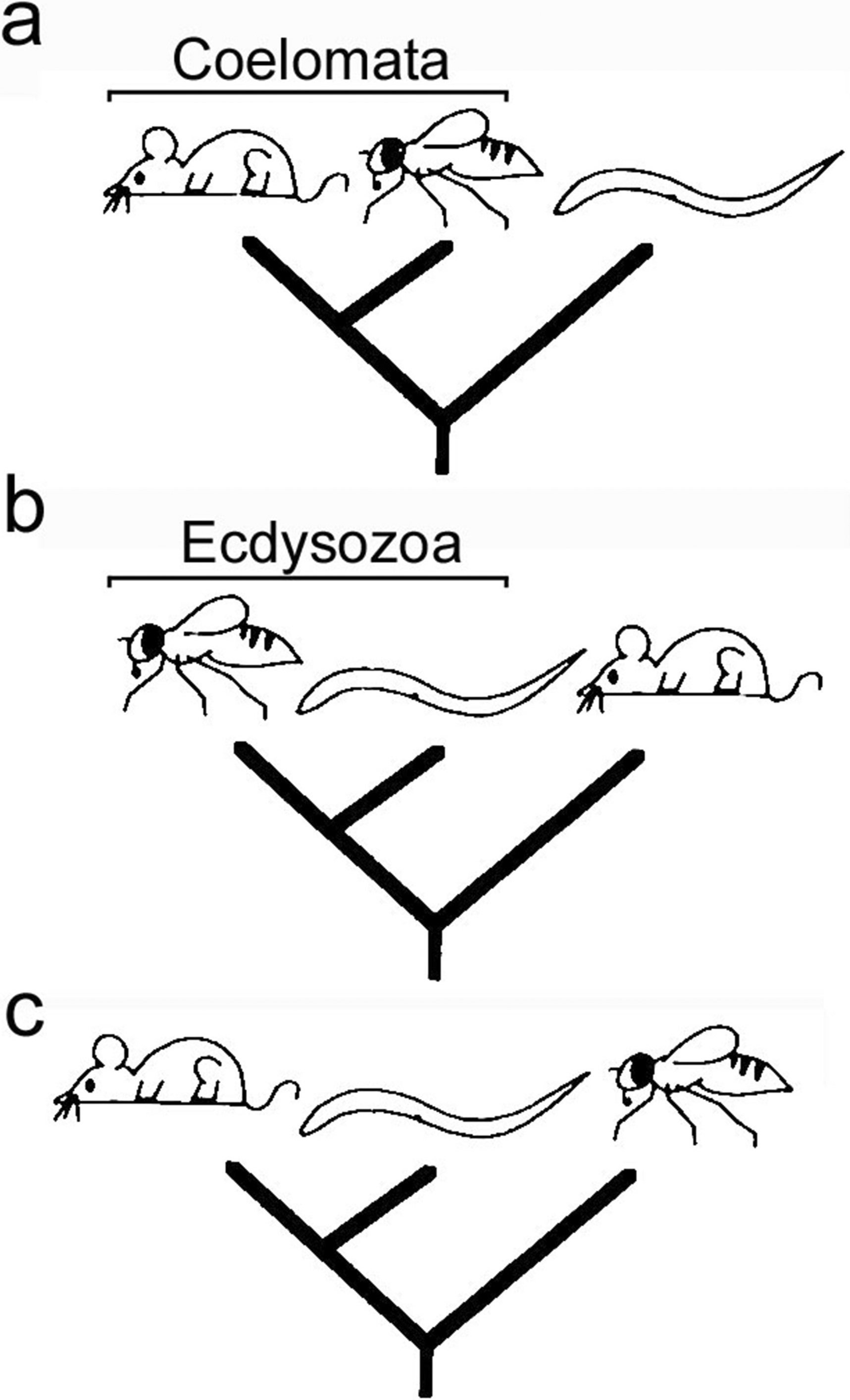
In C. elegans II, we outlined the 3 possible hypotheses (Figure 1) for relationships between three major model systems, "the" arthropod Drosophila melanogaster, "the" vertebrate Mus musculus, and "the" nematode C. elegans (Fitch and Thomas, 1997). We emphasized that elucidating these relationships was important for making inferences and predictions about which components, mechanisms and functions might be unique and derived or ancestrally shared by these or other related model and non-model species, such as humans. Of course, additional representatives of animals (including other nematodes) are needed in the phylogenetic framework for greater accuracy of such predictions. After we wrote that review, several interesting studies addressed relationships among the major animal phyla, and particularly the relationship of nematodes to other animals. There is still considerable (even polemical) debate, but additional data and increased analytical sophistication may provide answers in the not-distant future. As it is not possible here to review all of the work on animal phylogeny, and because the saga seems far from concluded, I highlight only the major findings relevant to the position of nematodes.
|
Figure 1. The three hypotheses for the relationships of three model organisms. (A) Mouse and fly are more closely related than either is to C. elegans, consistent with the Coelomata hypothesis; (B) Fly and worm are more closely related than either is to mouse, consistent with the Ecdysozoa hypothesis; (C) Worm and mouse are more closely related than either is to fly, not consistent with either of the major competing hypotheses of animal relationships. Modified from Fitch and Thomas (1997).
On the basis of complete 18S ribosomal RNA (rRNA) sequences, Aguinaldo et al., 1997 proposed that nematodes were related to arthropods in a clade of molting animals they called "Ecdysozoa" (Figure 1b), to the exclusion of deuterostomes (represented by an echinoderm in their study) and some other protostome groups, such as molluscs and annelids. This hypothesis differed substantially from the more traditional "Coelomata" hypothesis that placed nematodes on a branch diverging before coelomates diverged from one another (i.e., before the divergence of lineages leading to mice and flies, Figure 1a). Support for Ecdysozoa depended on excluding all but one nematode, Trichinella spiralis (which unfortunately possesses a rRNA sequence with an odd nucleotide composition compared to other nematodes characterized so far). However, when the authors included the other nematodes (e.g., C. elegans), the nematodes clustered together near the bottom of the tree, consistent with Coelomata, and consistent with data from RNA polymerase II (Sidow and Thomas, 1994). Use of Trichinella as a representative nematode was justified on the basis that its rRNA sequence evolved more slowly than that of other nematodes, such as C. elegans. A phenomenon called "long branch attraction" (LBA) can cause taxa with long branches (representing many evolutionary changes) to artefactually group with other long branches, particularly those of the outgroup taxa near the root of the tree (Felsenstein, 1978). Of course, the other possible reason that nematodes have long branches is simply that they diverged early from the other taxa, as predicted by the Coelomata hypothesis (Figure 2). The putative effect of LBA to provide artefactual support to Coelomata has been central to the debate, along with issues of taxon and character sampling.
|
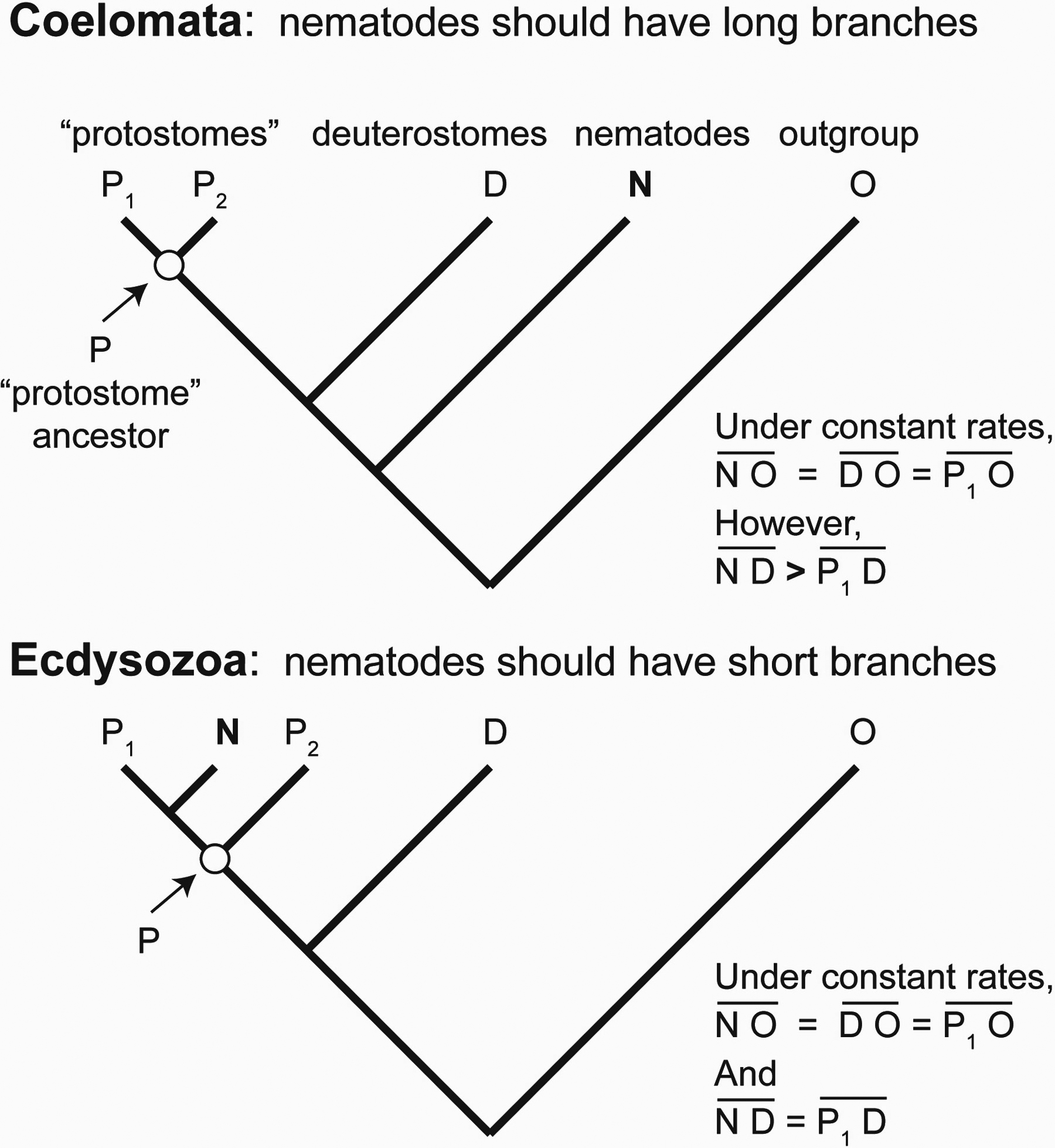
Figure 2. Why nematodes might have a long branch. It has been claimed that a phenomenon called "long-branch attraction" (LBA) results in an artefactual placement of nematodes near the base of the bilaterian phylogeny, thus appearing to be consistent with the "Coelomata" hypothesis, and obscuring the phylogenetic signal for "Ecdysozoa". However, the branch (lineage) leading to nematodes could be long for different reasons: (1) the rate of nucleotide change is higher in nematodes than in other taxa (e.g., Ecdysozoa could be true and support for Coelomata is artefactual), or (2) the nematodes branched earlier from the tree than the other taxa (i.e., Coelomata is true after all), or (3) both (i.e., Coelomata is true and the rate is higher in nematodes). One legitimate method for testing for significant LBA is to conduct a relative-rate test; one can then discard data (taxa or sequences) which show a significantly higher rate in nematodes than in other taxa. In the relative-rate test, the distance (amount of change, corrected for superimposed substitutions) between one ingroup taxon (like nematodes, N) and an outgroup (O, e.g. diploblastic animals, yeast, etc.) is compared to the distance between another ingroup taxon (e.g., a non-nematode protostome, P1, like Drosophila) and the same outgroup. If evolution has occured at equal rates in the different lineages, these distances, NO and P1O, should be approximately equal, regardless of which hypothesis (Coelomata or Ecdysozoa) is true. The outgroup, O, thus serves as an unbiased "anchor" for these rate comparisons. Any deviation from this equality is a measure of a rate difference. Data showing such a deviation may be discarded to see if their exclusion results in a change in the phylogenetic topology. It is not legitimate, however, to use a potential ingroup as an anchor for the relative-rate test. For example, if human (or other deuterostome taxon, D) were used, the relative rate test would be biased to favor Ecdysozoa. That is, although the distance between N and D should be the same as that between P1 and D if Ecdysozoa were true, ND should be greater than P1D if Coelomata were true and rates were equal. Eliminating data where ND> P1D would thus bias the selection of data to favor Ecdysozoa, even if Coelomata were true.
Unfortunately, the index that Aguinaldo et al., 1997, used to classify "long-branch taxa" was the distance between each taxon to the hypothetical last common ancestor of the protostomes (P in Figure 2), rather than to a more certain outgroup taxon, such as the cnidarian in their study, as is usually done in relative rate tests (Sarich and Wilson, 1967; Wu and Li, 1985; see Figure 2). That is, their method for selecting data to exclude assumed that nematodes diverged from other protostomes after deuterostomes diverged (as in the Ecdysozoa hypothesis). This assumption would be incorrect if Coelomata were true. Under the Coelomata hypothesis, the distance between nematodes and the hypothetical protostome ancestor is expected to be greater than the distance between arthropods and the protostome ancestor, even under constant evolutionary rates (Figure 2). Excluding taxa that show such a distance ratio would obviously be biased to favor Ecdysozoa. Instead, rates should be compared relative to a well-accepted outgroup taxon (O in Figure 2). Under the null hypothesis of constant rates, distances between any extant taxon and the outgroup should be the same; the amount of deviation from this equality can therefore be taken as the degree of rate difference (Figure 2).
The same problem applies to data selection in a study which found that 24 of 36 protein orthologs from Saccharomyces cerevisiae, C. elegans, Drosophila melanogaster, and human were consistent with Coelomata and only 11 were consistent with Ecdysozoa (Mushegian et al., 1998). The authors argued that LBA could result in artefactual support for Coelomata because most of the proteins supporting Coelomata had a longer branch between C. elegans and human than between Drosophila and human. However, this longer branch for C. elegans would be expected even if rates were perfectly clocklike and Coelomata were true (Figure 2). Unfortunately, this study was also hampered by the fact that most of their sets of putative "orthologs" were actually paralogs of various sorts, thus introducing another source of possible error for the inference of species relationships (Xie and Ding, 2000). Human was also used as the basis for comparing relative rates in a recent study which included genome-scale data for eleven taxa and reported strong support for Ecdysozoa when "long-branch" data were excluded (Dopazo and Dopazo, 2005). Thus, in all of the studies described above, inappropriate use of a potential ingroup taxon to measure relative rates could have mistakenly biased the conclusions in favor of Ecdysozoa.
It has been claimed that a phenomenon called "long-branch attraction" (LBA) results in an artefactual placement of nematodes near the base of the bilaterian phylogeny, thus appearing to be consistent with the "Coelomata" hypothesis, and obscuring the phylogenetic signal for "Ecdysozozoa" Figure 2.
Recent studies using genome-scale numbers of genes have generally supported Coelomata and rejected Ecdysozoa (e.g., Brown et al., 2001; Blair et al., 2002; Wolf et al., 2004). Some of these studies tested for effects of LBA and found no significant effect. The most recent study (Philip et al., 2005), used only single-copy genes from ten completely sequenced genomes to help ensure that only orthologs were compared and thus avoid complications of comparing potentially paralogous genes (which would have reflected gene duplication events and not the speciation events of interest). Their 780 orthologous genes, as well as the concatenated sequences of five genes meeting strict criteria for appropriate phylogenetic signal, strongly support Coelomata, even when sites with the highest evolutionary rates are removed. If support for Coelomata were merely due to an artifact of rapid evolution, removing these sites should produce support for Ecdysozoa. Removing additional sites with moderate rates results in ambiguity, but not support for Ecdysozoa.
A major criticism of these studies is that the sampling of taxa is small, as might be expected for whole-genome comparisons. To determine the effect of both taxon and character sampling, Philippe et al., (2005; reviewed by Jones and Blaxter, 2005) used data from 146 genes and a fairly diverse taxonomic sample of 35 species. In this case, the authors identified the fastest-evolving genes by appropriate comparison to the outgroup species and found strong support for Ecdysozoa when these genes were excluded. By including or removing taxa, the authors also demonstrated a clear effect of taxon selection. For example, adding hydra to the outgroup, which otherwise had only fungi and choanoflagellates, caused C. elegans to jump from a position consistent with Coelomata to one consistent with Ecdysozoa. The authors conclude that both accounting for LBA effects and including a denser sampling of taxa are required to uncover the phylogenetic signal for Ecdysozoa. This effect of taxon addition is explained by the ability of added taxa to "break" long branches and apportion changes more appropriately into different lineages, thus providing better phylogenetic information about which states are primitive and which are derived (Kim, 1998). Even when only one or a few genes are employed (such as for 18S rRNA), including more taxa has apparently aided resolution, generally resulting in support for Ecdysozoa (Giribet and Ribera, 1998; Giribet and Wheeler, 1999; Peterson and Eernisse, 2001). Adding more taxa, however, means that statistically testing the robustness of relationships becomes computationally much more time-consuming and most of these taxon-dense analyses do not have such tests.
In this brief review, I have focused on contributions from recent work using multigene data. However, insight might also be provided by a few molecular characters which nevertheless have phylogenetic signatures complex enough to have arisen only once, such as insertions, deletions, and organization of gene clusters. For example, Hox gene organization may be one such complex—and therefore informative—feature (Balavoine et al., 2002). The Abd-B gene appears to be specific for Ecdysozoa. However, orthology of the C. elegans php-3 to Abd-B is only very weakly supported (42% bootstrap value; de Rosa et al., 1999). One problem with C. elegans as a representative nematode is that it clearly has a highly derived organization of the Hox gene cluster relative to other nematodes; genes have been lost and rearrangements have occurred in lineages leading to C. elegans (Aboobaker and Blaxter, 2003). Analyses using other nematode species should therefore provide better phylogenetic information than C. elegans for addressing the Ecdysozoa/Coelomata question. Other potentially informative molecular characters are insertions, deletions and intragenic rearrangements. In this case, Coelomata appears to be supported (Wolf et al., 2004). On the other hand, intron patterns seem to support Ecdysozoa. In an essentially 4-taxon study, Roy and Gilbert, 2005 found that patterns of "intron conservation" (actually shared losses of introns) tended to support Ecdysozoa, under the assumption that introns were never regained in the same position. For all of these studies, the problem of low taxon density could be problematic. The extreme dynamic of intron loss and gain even within one genus of nematodes, Caenorhabditis (Kiontke et al., 2004), suggests one should be wary of assigning homologies to intron presence or absence in taxa as distantly related as different phyla.
There are still uncertainties and questions. Because of improvements caused by better taxon sampling, adding more taxa that represent more animal phyla could potentially alter the tree topology again. Because Ecdysozoa predicts that "molting animals" shared a common ancestor that was itself a molting animal, we might expect that molting in C. elegans and Drosophila share common mechanisms (although the cuticles that are molted in these animals are very different). In fact, orthologs of some of the nuclear hormone receptors (NRs) involved in molting and metamorphosis in insects also function in growth and dauer molts in C. elegans (Gissendanner et al., 2004). The filarial nematode, Dirofilaria immitis, has putative orthologs of the ecdysone receptor gene and other ecdysone-regulated genes that are absent from the C. elegans genome (Crossgrove et al., 2002; Shea et al., 2004). Of course, filaria are parasites of insects, and a means to respond to insect hormones would have been adaptive and could have evolved secondarily. An even more important issue is whether these NR genes or hormone pathways could have been shared by much more ancient common ancestry and lost or modified in other lineages. In fact, ecdysteroids are found throughout eukaryotes. Such features may not represent derived changes ("apomorphies") supporting Ecdysozoa. As an analogy, just because humans and rats have four limbs does not mean that four limbs supports a close relationship between humans and rats, since four limbs are shared ("plesiomorphically") by all tetrapods (except where they have been lost secondarily, as in snakes). Cuticular shedding also occurs in non-ecdysozoan phyla, such as in some annelids and sipunculans (Brusca and Brusca, 2003), so even the nominal feature of Ecdysozoa cannot be used exclusively to support this taxon.
Even if Ecdysozoa turns out to be the most well-supported hypothesis at the molecular level, there are many anatomical features of nematodes that will have to be explained (see Nielsen, 2001; Brusca and Brusca, 2003). Several features presumably present in an ecdysozoan ancestor must have been lost or reduced and/or completely remodeled in nematodes (appendages, segmentation, excretory system, nervous system). Although it is conceivable that coeloms have been reduced to "pseudocoeloms" several times during animal evolution by heterochrony or similar means and that specialized gas exchange systems were eliminated as a response to the evolution of small body size, it is difficult to envisage how nematodes could have lost such features as bilateral ganglia after they had evolved in an ancestor (Brusca and Brusca, 2003). As more genomes are sequenced for more representatives of different animal phyla, and the hypotheses are appropriately tested with the increasingly sophisticated tools of phylogenetics, the position of nematodes in the Tree of Life will become clearer.
Many thanks to Thomas Bürglin for stimulating discussions about the Ecdysozoa debate and to Mark Blaxter, Rob DeSalle, Gonzalo Giribet, and Karin Kiontke for their valuable comments on the manuscript.
Aboobaker, A.A., and Blaxter, M.L. (2003). Hox gene loss during dynamic evolution of the nematode cluster. Curr. Biol. 13, 37–40. Abstract Article
Aguinaldo, A.M., Turbeville, J.M., Linford, L.S., Rivera, M.C., Garey, J.R., Raff, R.A., and Lake, J.A. (1997). Evidence for a clade of nematodes, arthropods and other molting animals. Nature 387, 489–493. Abstract Article
Balavoine, G., de Rosa, R., and Adoutte, A. (2002). Hox clusters and bilaterian phylogeny. Mol. Phylogenet. Evol. 24, 366–373. Abstract Article
Blair, J.E., Ikeo, K., Gojobori, T., and Hedges, S.B. (2002). The evolutionary position of nematodes. BMC Evol. Biol. 2, 7. Abstract Article
Brown, J.R., Douady, C.J., Italia, M.J., Marshall, W.E., and Stanhope, M.J. (2001). Universal trees based on large combined protein sequence data sets. Nat. Genet. 28, 281–285. Abstract Article
Brusca, R.C., and Brusca, G.J. (2003). Invertebrates, 2nd edn (Sunderland, MA: Sinauer Associates), 936 p.
Crossgrove, K., Laudet, V., and Maina, C.V. (2002). Dirofilaria immitis encodes Di-nhr-7, a putative orthologue of the Drosophila ecdysone-regulated E78 gene. Mol. Biochem. Parasitol. 119, 169–177. Abstract Article
de Rosa, R., Grenier, J.K., Andreeva, T., Cook, C.E., Adoutte, A., Akam, M., Carroll, S.B., Balavoine, G. (1999). Hox genes in brachiopods and priapulids and protostome evolution. Nature 399, 772–776. Abstract Article
Dopazo, H., and Dopazo, J. (2005). Genome-scale evidence of the nematode-arthropod clade. Genome Biol. 6, R41. Abstract Article
Eisen, J.A. (1998). Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 8, 163–167. Abstract
Eisen, J.A., and Fraser, C.M. (2003). Phylogenomics: intersection of evolution and genomics. Science 300, 1706–1707. Abstract Article
Felsenstein, J. (1978). Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 27, 401–410.
Fitch, D.H.A., and Thomas, W.K. (1997). Evolution. In C. elegans II, D.L. Riddle, T. Blumenthal, B.J. Meyer, and J.R. Priess, eds. (Cold Spring Harbor: Cold Spring Harbor Laboratory Press), pp. 815–850.
Giribet, G., and Ribera, C. (1998). The position of arthropods in the animal kingdom: a search for a reliable outgroup for internal arthropod phylogeny. Mol. Phylogenet. Evol. 9, 481–488. Abstract Article
Giribet, G., and Wheeler, W.C. (1999). The position of arthropods in the animal kingdom: Ecdysozoa, islands, trees, and the "Parsimony ratchet". Mol. Phylogenet. Evol. 3, 619–623. Abstract Article
Gissendanner, C.R., Crossgrove, K., Kraus, K.A., Maina, C.V., and Sluder, A.E. (2004). Expression and function of conserved nuclear receptor genes in Caenorhabditis elegans. Dev. Biol. 266, 399–416. Abstract Article
Jones, M., and Blaxter, M. (2005). Evolutionary biology: animal roots and shoots. Nature 434, 1076–1077. Abstract Article
Kim, J. (1998). Large-scale phylogenies and measuring the performance of phylogenetic estimators. Syst. Biol. 47, 43–60. Abstract Article
Kiontke, K., Gavin, N.P., Raynes, Y., Roehrig, C., Piano, F., and Fitch, D.H.A. (2004). Caenorhabditis phylogeny predicts convergence of hermaphroditism and extensive intron loss. Proc. Natl. Acad. Sci. USA 101, 9003–9008. Abstract Article
Mushegian, A.R., Garey, J.R., Martin, J., and Liu, L.X. (1998). Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 8, 590–598. Abstract
Nielsen, C. (2001). Animal Evolution: Interrelationships of the Living Phyla, 2nd edn (Oxford: Oxford University Press).
Peterson, K.J., and Eernisse, D.J. (2001). Animal phylogeny and the ancestry of bilaterians: inferences from morphology and 18S rDNA gene sequences. Evol. Dev. 3, 170–205. Abstract Article
Philip, G.K., Creevey, C.J., and McInerney, J.O. (2005). The opisthokonta and the ecdysozoa may not be clades: stronger support for the grouping of plant and animal than for animal and fungi and stronger support for the coelomata than ecdysozoa. Mol. Biol. Evol. 22, 1175–1184. Abstract Article
Philippe, H., Lartillot, N., and Brinkmann, H. (2005). Multigene analyses of bilaterian animals corroborate the monophyly of ecdysozoa, lophotrochozoa, and protostomia. Mol. Biol. Evol. 22, 1246–1253. Abstract Article
Roy, S.W., and Gilbert, W. (2005). Resolution of a deep animal divergence by the pattern of intron conservation. Proc. Natl. Acad. Sci. USA 102, 4403–4408. Abstract Article
Sarich, V.M., and Wilson, A.C. (1967). Immunological time scale for hominid evolution. Science 158, 1200–1203. Abstract
Shea, C., Hough, D., Xiao, J., Tzertzinis, G., and Maina, C.V. (2004). An rxr/usp homolog from the parasitic nematode, Dirofilaria immitis. Gene 324, 171–182. Abstract Article
Sidow, A., and Thomas, W.K. (1994). A molecular evolutionary framework for eukaryotic model organisms. Curr. Biol. 4, 596–603. Abstract Article
Sudhaus, W., and Fitch, D. (2001). Comparative studies on the phylogeny and systematics of the Rhabditidae (Nematoda). J. Nematol. 33, 1–70.
Winnepenninckx, B., Backeljau, T., Mackey, L.Y., Brooks, J.M., De Wachter, R., Kumar, S., and Garey, J.R. (1995). 18S rRNA data indicate that Aschelminthes are polyphyletic in origin and consist of at least three distinct clades. Mol. Biol. Evol. 12, 1132–1137.
Wolf, Y.I., Rogozin, I.B., and Koonin, E.V. (2004). Coelomata and not Ecdysozoa: evidence from genome-wide phylogenetic analysis. Genome Res. 14, 29–36. Abstract Article
Wu, C.-I, and Li, W.-H. (1985). Evidence for higher rates of nucleotide substitution in rodents than in man. Proc. Natl. Acad. Sci. USA 82, 1741–1745. Abstract
*Edited by Lisa R. Girard. Last revised June 08, 2005. Published August 31, 2005. This chapter should be cited as: Fitch, D.H.A., Introduction to nematode evolution and ecology (August 31, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.19.1, http://www.wormbook.org.
Copyright: © 2005 David H.A. Fitch. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: david.fitch@nyu.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.