Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergAbstract
The mitochondrial genome is vital for Caenorhabditis elegans metabolism, physiology, and development. The C. elegans mitochondrial DNA is typical of animal mitochondrial genomes in its size and gene content. It is 13,794 nucleotides in length and encodes 36 genes: 2 ribosomal RNAs, 22 transfer RNAs, and 12 protein subunits of the mitochondrial respiratory chain. Although it represents only a small number of genes, an elaborate cellular machinery comprised of over 200 nuclear genes is needed to replicate, transcribe, and maintain the mitochondrial chromosome and to assemble the translation machinery needed to express this dozen proteins. Mitochondrial genetics is peculiar and complex because mitochondrial DNA is maternally inherited and can be present at tens to tens of thousands of copies per cell. The mitochondrial genome content of the developing nematode is developmentally regulated; it increases about 30-fold between the L1 and the adult stages and blocking the increase leads to larval arrest. Energy metabolism is also intimately linked to aging and lifespan determination. The nematode model system offers numerous advantages for understanding the full importance and scope of the mitochondrial genome in animal life.
The mitochondrial genome is indispensable to the cellular and organismal biology of Caenorhabditis elegans. An elaborate cellular machinery is employed to maintain mitochondrial DNA (mtDNA), to express it, and to ensure its inheritance. Because it is maternally transmitted and because of its wide-ranging copy number (it can be present at fewer than 100 to tens of thousands of copies per cell), the genetics of mtDNA are characterized by a number of peculiar and as yet incompletely understood features. The fundamental importance of mtDNA to cellular energy metabolism explains the profound effects mtDNA mutations can have; mitochondria are the major source of reactive oxygen species, which are implicated in premature aging and senescence.
Mitochondria play a pivotal role in cellular metabolism. They were once free-living organisms related to modern eubacteria and they continue to perform many of the biochemical and physiological functions of their bacterial ancestors (Timmis et al., 2004). Mitochondria are double-membrane organelles most commonly associated with oxidative phosphorylation, a process that meets the majority of cellular energy demands. In addition, mitochondria are involved in heme, lipid, nucleotide, iron-sulfur cluster, and amino acid biosynthesis: they are home to the citric acid cycle, the urea cycle and fatty acid oxidation.
Mitochondria harbor a small but essential component of an eukaryote's genetic material. The symbiotic relationship that marks the origin of eukaryotes was established approximately 2 billion years ago and has been followed by a massive loss or transfer of genes to the host genome; only a tiny fraction (less than one percent) of the endosymbiont's genome is retained on the mtDNA (Gray et al., 2001). Animal mtDNAs are extremely compact, being less than 20 kilobases in length, and encode fewer than 40 genes (Wolstenholme, 1992). All of the proteins encoded by the mtDNA are subunits of the mitochondrial respiratory chain (MRC; Okimoto et al., 1992). Despite their small number, the genes on the mtDNA necessitate a large expenditure of cellular resources because they are essential for strict aerobes. Hundreds of nuclear genes are needed to replicate, transcribe, and maintain the mitochondrial chromosome and to assemble the translation machinery needed to express its dozen or so proteins (Tsang and Lemire, 2003).
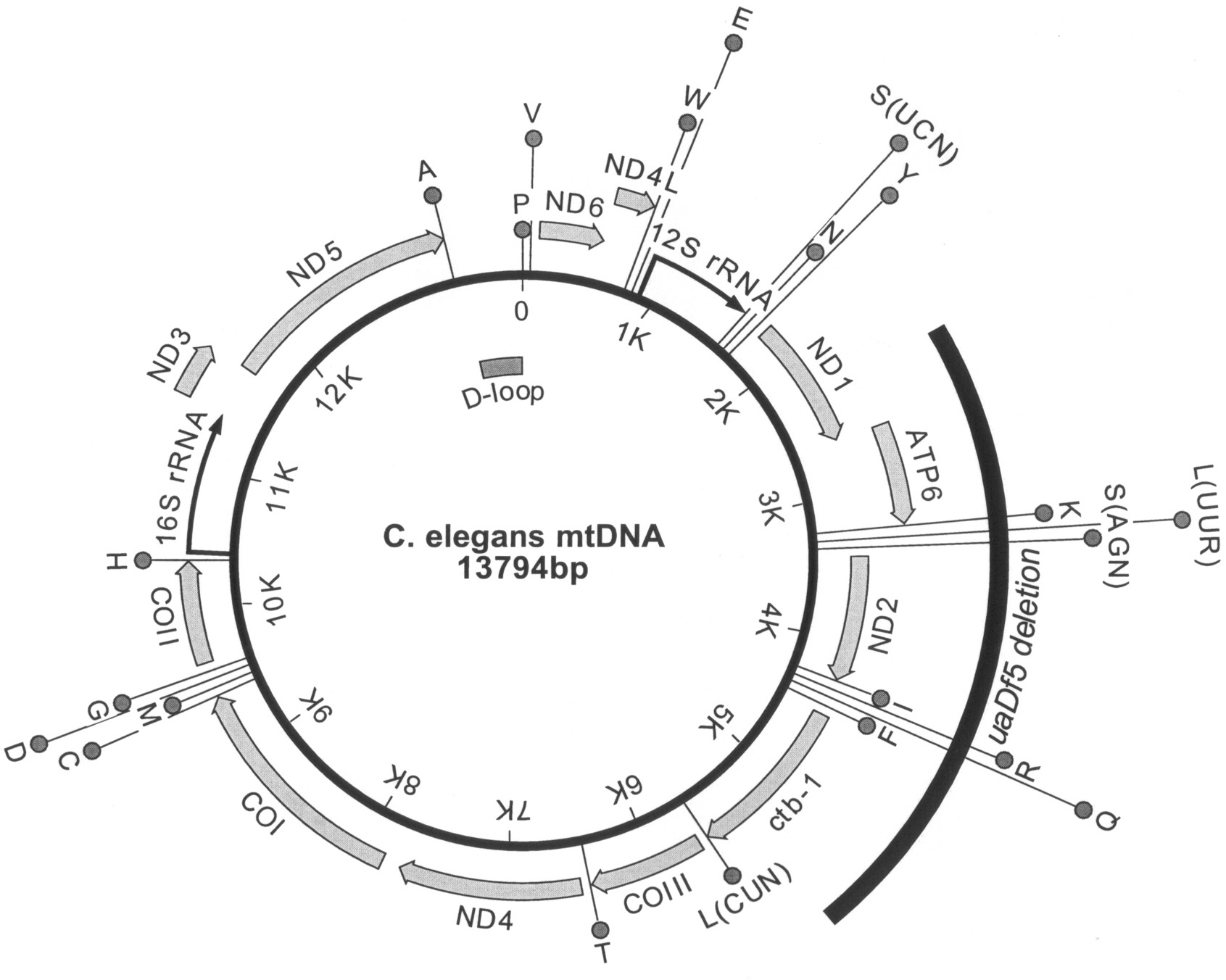
The C. elegans mtDNA is 13,794 nucleotides in length and encodes 36 genes: 2 ribosomal RNAs (12S rRNA and 16S rRNA), 22 transfer RNAs, and 12 MRC subunits (Figure 1; Okimoto et al., 1992). A typical animal mtDNA, the nematode mitochondrial genome is slightly smaller than its human counterpart. It lacks the ATP8 gene of the human genome, which encodes a subunit of the ATP synthase (complex V). Introns are absent and there are few or no non-coding nucleotides between genes; approximately 92% of the mitochondrial genome has coding function. Three genes end with incomplete termination codons (T or TA) that are apparently converted to TAA codons by polyadenylation of the transcript. Only one sizeable non-coding region called the displacement loop or D-loop is present. The replication and transcription of mtDNA has been mainly studied in mammals where the D-loop region contains one of the origins of replication and the promoters for mtDNA transcription (Garesse and Vallejo, 2001). Few components needed for the maintenance or expression of the C. elegans mtDNA have been experimentally identified. The CLK-1 (biological clock abnormal) protein was reported to have DNA binding activity specific for the light strand origin of replication (OL) on the mtDNA, suggesting a role in regulating mtDNA replication or transcription in C. elegans (González-Halphen et al., 2004; Gorbunova and Seluanov, 2002).
|
Figure 1. Gene map of the C. elegans mtDNA. The molecule contains the genes for twelve proteins (thick grey arrows), two rRNAs (black arrows), and 22 tRNAs (circles labeled with one-letter amino acid code). The serine and leucine tRNAs are also identified by the codon family recognized. The positions of the putative D-loop and of the uaDf5 deletion mutation are indicated inside and outside the circle, respectively.
In contrast to the relatively uniform gene content of metazoan mtDNAs, the mitochondrial genetic codes are highly modified. In C. elegans, TGA specifies tryptophan rather than being a stop codon, ATA specifies methionine rather than iso-leucine, and AGA and AGG specify serine rather than arginine. The translation initiation codon ATG is not used; ATT, ATA, or TTG codons apparently serve as initiation codons (Okimoto et al., 1992; Okimoto et al., 1990). These peculiarities of the genetic code may in part explain the retention of the twelve protein-coding genes on the mtDNA rather than their transfer to the nucleus.
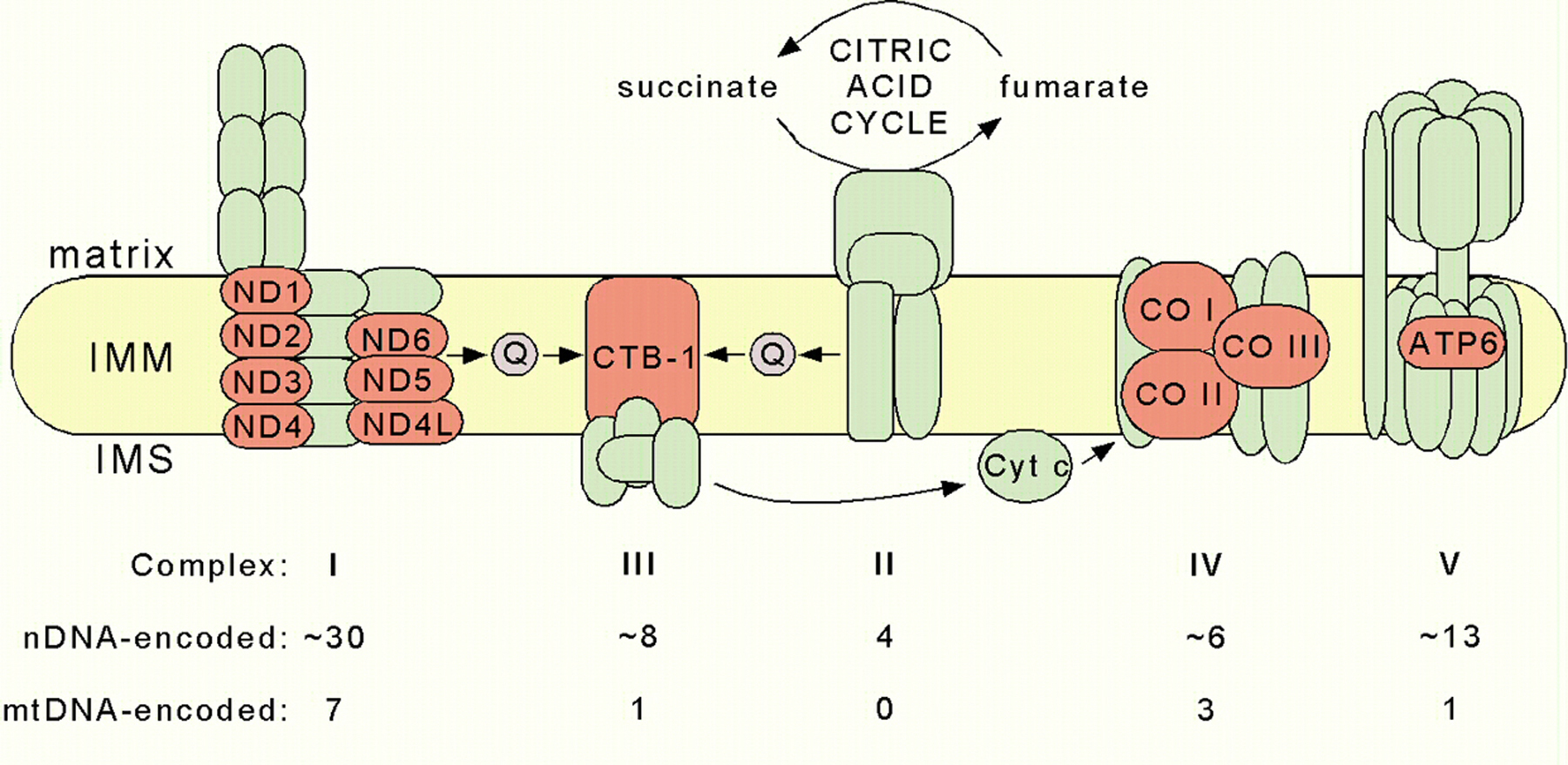
The C. elegans mtDNA encodes 12 MRC subunits out of a total of the approximately 80 subunits assembled into five MRC complexes (Figure 2). Four of these five complexes contain mtDNA-encoded subunits; complex II, the succinate-ubiquinone oxidoreductase complex, is the exception. The ND1, ND2, ND3, ND4, ND4L, ND5, and ND6 subunits of complex I (the NADH-ubiquinone oxidoreductase) are all core subunits localized in the membrane arm of the enzyme. The ND1 subunit is implicated in ubiquinone binding (Schultz and Chan, 2001). The cytochrome b of complex III (the ubiquinol-cytochrome c oxidoreductase) is encoded by the ctb-1 gene in C. elegans; it is an essential component of the enzyme, harboring 2 b-type hemes. Similarly, the COI, COII, and COIII subunits, which coordinate catalytic heme and copper cofactors, are essential components of complex IV (the cytochrome c oxidase). Finally, the ATP6 subunit of complex V plays a vital role in the efficiency of ATP synthesis (Lenaz et al., 2004). Not surprisingly, mutations in each of the human orthologs are causal in a variety of neurological, endocrinological, and muscular diseases (DiMauro, 2004).
|
Figure 2. Schematic representation of the mitochondrial respiratory chain. The twelve mtDNA-encoded subunits in C. elegans are shown in red and nuclear DNA (nDNA)-encoded subunits in green. The table indicates the estimated numbers of subunits that comprise the nematode complexes. IMM, inner mitochondrial membrane; IMS, intermembrane space; Q, ubiquinone; Cyt c, cytochrome c.
Very few mutations in the nematode mtDNA have been reported. In contrast, over 100 point mutations and 200 insertions, deletions, or rearrangements have been described for the human mtDNA (see MITOMAP: A Human Mitochondrial Genome Database). It is puzzling why mtDNA mutations have not been reported in C. elegans, given the large number of genetic screens for defects in motility, reproduction, development, and other functions. Perhaps the nematode can better tolerate deleterious mtDNA mutations or has mechanisms to prevent their transmission. The genetics of the mitochondrial genome still offer much to be explored.
It is generally believed that animal mtDNA is inherited exclusively from the mother. However, it was recently shown in a patient with mitochondrial myopathy that the pathogenic mtDNA was of paternal origin (Schwartz and Vissing, 2002). Evidence for the occasional paternal transmission of mtDNA has also been reported in sheep, mice, cattle, mussels, and insects (Zhao et al., 2004). Although rare, the paternal inheritance of mtDNA may have a significant impact on evolution and on disease development.
Mitochondria are dynamic structures and organelle plasticity is related to mtDNA inheritance (Garrido et al., 2003). Multiple mtDNA molecules are organized into discrete protein-DNA complexes called nucleoids (Jacobs et al., 2000). Mitochondria form reticular or tubular networks and their interaction with cytoskeletal components provides clues to their distribution, movement, and inheritance (Rube and van der Bliek, 2004). A mutation in the anc-1 (anchorage of nuclei abnormal) gene of C. elegans resulted in hypodermal nuclei and mitochondria that float freely in the cytoplasm of syncytial cells (Hedgecock and Thomson, 1982). These mitochondria adopted an almost spherical shape and were often clustered. ANC-1 contains several coiled coil regions, a nuclear envelope localization domain, and an actin-binding domain, suggesting it mediates connections from nuclei and mitochondria to the actin network and to the nuclear envelope (Starr and Han, 2002). Mitochondrial division is required to regulate organelle numbers during cell division, differentiation, and in response to environmental conditions. The C. elegans DRP-1, a dynamin-related protein with a characteristic GTPase domain, plays a key role in mitochondrial distribution and division (Labrousse et al., 1999). RNAi of drp-1 results in embryonic lethality and abnormal mitochondrial morphology and distribution, while overexpression leads to fragmentation of the organelle. DRP-1 is involved in the fission of the mitochondrial outer membrane, perhaps directly or perhaps through its ability to recruit additional fission machinery components.
The numbers of mitochondria and mitochondrial genomes in a cell are regulated and can vary substantially between tissues (Moraes, 2001). The developmental regulation of mtDNA copy numbers in C. elegans has been investigated (Tsang and Lemire, 2002). An embryo contains ∼25,000 copies of mtDNA and this number remains unchanged through the L1, L2, and L3 larval stages. A fivefold increase to 1.3 x 105 copies in the L4 stage is followed by a further sixfold increase to 7.8 x 105 copies in the adult hermaphrodite. The first copy number increase coincides with sexual maturation; spermatogenesis begins in the early L4 stage and oogenesis in the young adult.
mtDNA copy number is closely tied to reproduction. Mutations in the glp-1 gene, which is involved in germ line proliferation, lead to an almost complete absence of germ line cells while somatic cell numbers are normal (Austin and Kimble, 1987). The mtDNA content of an L4-stage glp-1 hermaphrodite is ∼70,000, half of wild type levels (Tsang and Lemire, 2002). There is no further increase in mtDNA copy number as the glp-1 animal matures. Thus, increases in mtDNA content from the L3 to the adult stages can be subdivided into two components: a somatic component is glp-1 independent and occurs during development from the L3 (∼25,000 copies) to the L4 stage (∼70,000 in the glp-1 L4) and a germ line component accounts for the remainder of the increase to 7.8 x105 in the adult.
Oocyte production accounts for the majority of the germ line-related increases in mtDNA copy numbers. A loss of function mutation in the feminization gene fem-1 blocks sperm production in the hermaphrodite but does not affect mtDNA contents in the L4 or in the adult (Tsang and Lemire, 2002). In contrast, a gain of function mutation in the fem-3 gene, which blocks oocyte production, does not affect L4 mtDNA content but reduces the content in the adult to about one quarter (to 1.9 x 105 copies) of wild type levels. Thus, the majority of organellar genomes are associated with oocyte development and the sperm-associated component is minor. When determined independently, 18,000 copies of mtDNA were measured per fem-1 oocyte, a number similar to wild type embryos and early larvae.
The mtDNA content of individual C. elegans cells is considerably lower than the estimated values of 1,000-10,000 copies per cell in higher eukaryotes (Tsang and Lemire, 2002). The ∼25,000 copies of mtDNA in the L1 larva, if evenly distributed around the 558 nuclei, suggest an average of 45 copies per cell. Similarly, the L4 larva has ∼1,000 somatic nuclei and 70,000 copies of mtDNA in a glp-1 mutant, corresponding to 70 copies per cell. It remains to be determined whether different somatic cells or tissues have distinct mitochondrial genome contents that might reflect their energy requirements. Estimated cellular contents of mtDNA for undifferentiated germ line cells and for sperm are 250 and 30-40, respectively. The increases in mtDNA copy numbers during development are suggestive of parallel increases in the numbers of organelles, although this has not been documented. Interestingly, there is an increase in organelle number and in the frequency of organelle division with a shift of temperature from 15 to 25°C (Labrousse et al., 1999). This increased content of mitochondria may reflect an adaptation to the higher metabolic demands of elevated temperatures. Consistent with this notion, respiration rates (Van Voorhies and Ward, 1999) and ubiquinone levels (Jonassen et al., 2002) are doubled with similar temperature shifts.
A functional MRC is essential for viability and larval development past the L3 stage. Mutations in nuclear genes encoding MRC subunits such as nuo-1 or atp-2 (subunits of complexes I and V respectively) lead to a characteristic L3 stage arrest having an L2-staged gonad (Tsang and Lemire, 2003; Tsang et al., 2001). Similarly, clk-1 mutations, which impair ubiquinone biosynthesis, can lead to an L2 or L3-stage arrest (Hihi et al., 2002; Jonassen et al., 2001). Inhibitors of mtDNA replication or transcription such as ethidium bromide or inhibitors of mitochondrial translation such as chloramphenicol or doxycycline can also quantitatively produce an L3 stage developmental arrest (Tsang and Lemire, 2002; Tsang et al., 2001). Maturation to the L4 stage likely entails increased energy demands that are met with de novo synthesis of new mitochondrial energy production capacity. An energy sensor that responds to the concentrations of one or more metabolites, such as ATP or NADH, may communicate information about the status of mitochondrial energy generation and lead to altered patterns of MRC gene expression (Tsang and Lemire, 2003; Tsang et al., 2001).
Mitochondrial genetics differ from nuclear genetics in three aspects. First, mitochondrial genes are maternally inherited; they do not follow a Mendelian pattern of inheritance. Second, the mitochondrial genome is polyploid. Normally, a state of homoplasmy exists where only one form of mtDNA is present. Mutation can lead to a state of heteroplasmy where two or more forms of mtDNA coexist within a cell. Third, unlike a diploid nuclear gene that can normally only assume three states (homozygous wild type, heterozygous, or homozygous mutant), mtDNA heteroplasmy does not vary by discrete steps. The proportions of mtDNA species can vary with time or through mitotic segregation as cells divide.
The inheritance and maintenance of mtDNA species are most poorly understood in the heteroplasmic condition. The uaDf5 mtDNA deletion, which removes 11 genes (four MRC subunit and seven tRNA genes), is maternally inherited and stably propagated without selection (Figure 1; Tsang and Lemire, 2002). While individual animals have uaDf5 mtDNA contents ranging from ∼20% to ∼80%, the population average is ∼60% and this value does not vary between larval stages. There is no phenotype associated with the deletion, even in animals with an ∼80% content of uaDf5 mtDNA. Most surprisingly, homoplasmic wild type or mutant animals have not been detected in over 100 generations, suggesting the two forms of mtDNA do not segregate from each other. Single, self-fertilized hermaphrodites with intermediate levels of uaDf5 mtDNA (∼50-60%) have offspring containing from 20-80% mutant mtDNA, indicating that the level of heteroplasmy can change significantly in one generation. However, hermaphrodites with extreme levels of heteroplasmy (∼20% or ∼80%) only produce offspring with more moderate levels, suggesting two opposing forces are in operation. One force increases uaDf5 contents when they are low, while the second decreases them when they are high. One possible explanation for this phenomenon is that wild type mtDNA contains an undetected mutation that is complemented by the uaDf5 mtDNA. Thus, both genomes are needed to produce a fully functional MRC and homoplasmy for either mutation would be severely compromising or lethal. Alternatively, the stable heteroplasmy may result from intra-mitochondrial heteroplasmy, a condition where the mtDNA species are intermixed within the nucleoid of one organelle (Jacobs et al., 2000).
Although the heteroplasmic uaDf5 animals do not have a phenotype, they have adjusted their mtDNA contents. Both hermaphrodites and males have approximately twice the number of mitochondrial genomes as their wild type counterparts (Tsang and Lemire, 2002). The upregulation of mtDNA copy number may in part compensate for the effects of the mutation by enhancing the production of functional mitochondrial transcripts and/or proteins. Understanding the signals and mechanisms employed to regulate mtDNA copy number remains an important challenge.
The C. elegans mtDNA mutation rate has been estimated using a series of mutation accumulation lines maintained by single-progeny descent for over 200 generations (Denver et al., 2000). In over 770,000 base pairs of sequenced DNA, 16 base substitution and 10 insertion/deletion mutations were detected, corresponding to a measured rate approximately 100-fold higher than previous indirect estimates. Four of the insertion/deletion mutations introduced drastic changes to coding sequences, were homoplasmic (or nearly so), but had only modest effects on fitness. Most spontaneous mtDNA mutations in C. elegans appear to be mildly deleterious although selection may occur in a natural context.
The mtDNA mutation rate can be increased by environmental agents or by mutation of nuclear genes involved in mtDNA maintenance. For example, nucleoside analogs used in the clinical treatment of viral infections, such as human immunodeficiency virus, inhibit the mtDNA polymerase, which is essential for mtDNA synthesis and repair (Martin et al., 2003). A mutation that impairs the fidelity of the mtDNA polymerase produced a mtDNA mutator phenotype in a mouse model; this was associated with premature aging and reduced lifespan (Trifunovic et al., 2004).
Mitochondrial functions influence and possibly control the rate of aging (Dillin et al., 2002) and conversely, aging affects the integrity of the mitochondrial genome. An increased production of reactive oxygen species, often associated with MRC dysfunction, can precipitate premature aging, cause mutations in the mitochondrial and nuclear genomes, and shorten lifespan (Hartman et al., 2004). The first C. elegans mtDNA deletions detected were spontaneous events in an aging, wild type population (Melov et al., 1994; Melov et al., 1995). age-1 (aging abnormal) mutants, which are more resistant to oxidative stress and live twice as long as wild type, have a lower frequency of mtDNA deletions (Melov et al., 1995). It has not been determined whether these aging-associated mtDNA deletions reduce fitness or whether they are heritable.
To date, there is no direct evidence linking genes encoded by mtDNA with aging, but mutations in nuclear genes provide ample reason to believe that they are involved. The isp-1(qm150) mutation, which affects the iron sulfur protein of complex III, was the first mutation of an MRC subunit reported to increase lifespan (Feng et al., 2001). The mtDNA-localized, maternally-inherited ctb-1(qm189) mutation affecting the cytochrome b subunit of complex III, does not alter the long life span of qm150 animals but does partially suppress other effects of the qm150 mutation (Feng et al., 2001). The ctb-1(qm189) mutation has no independent phenotype. The genetic interaction of the isp-1 and ctb-1 mutations clearly illustrates the interplay of the mitochondrial and nuclear genomes in cellular metabolism. The mev-1(kn1) mutation is a missense allele of the cyt-1 gene encoding the cytochrome b subunit of complex II. Mutants are hypersensitive to methyl viologen and oxidative stress, have elevated levels of superoxide anion production, display signs of premature aging, have a reduced lifespan, and are hypermutable (Hartman et al., 2001; Ishii et al., 1998; Ishii et al., 2002; Senoo-Matsuda et al., 2001). A probable null mutation in the gene for the mitochondrial leucyl-tRNA synthetase (lrs-2(mg312)) markedly extends life span (Lee et al., 2003). The synthetase charges mitochondrial tRNAs needed for the translation of the 12 mtDNA-encoded MRC subunits. Mutation of the gene encoding the isopentenyl-pyrophosphate:tRNA transferase (gro-1(e2400), abnormal growth rate), produces a Clk (biological clock abnormal) phenotype characterized by slow development and increased lifespan (Lemieux et al., 2001). The apparent use of alternative translation initiation codons produces differentially targeted forms of GRO-1; it is the loss of the mitochondria-targeted form, which presumably modifies a subset of tRNAs and affects mitochondrial translation, that leads to the Clk phenotype.
Non-genetic interventions that affect mitochondrial function can also affect aging. Systematic RNAi screens for increased lifespan have identified numerous mitochondrial components, including MRC subunits, mitochondrial carrier proteins, a subunit of the mitochondrial ribosome, and a protein involved in MRC biogenesis (Dillin et al., 2002; Lee et al., 2003). Mutations in the clk-1 gene disrupt ubiquinone biosynthesis and significantly increase lifespan (Ewbank et al., 1997; Wong et al., 1995). Finally, exposure to antimycin A, a tight-binding inhibitor of complex III, reduces rates of respiration and promotes longevity (Dillin et al., 2002). Given the presence of 12 key MRC genes on the mtDNA and the intimate connections between MRC function and lifespan, it seems inevitable that mutations that slow aging will be identified in one or more of those 12 genes.
The mitochondrial genome is often ignored although it is a vital component of animal metabolism, physiology, and development. Few studies have specifically addressed the C. elegans mtDNA and much of our knowledge is inferred from investigations of vertebrate mtDNAs and from bioinformatic approaches. The C. elegans cellular machinery required for mtDNA maintenance and expression is almost completely unexplored; undoubtedly biological novelties remain to be exposed. Deciphering how mitochondria communicate with the remainder of the cell will have far-reaching implications for understanding cellular metabolism in normal and in diseased states. Few model systems offer as many advantages as the nematode for grasping the full importance and scope of mitochondrial genetics in animal life.
I wish to thank Catherine McPhalen for critical reading of the manuscript. The Canadian Institutes of Health Research Grant MT-15336 supported this work. Bernard Lemire is a member of the Membrane Protein Research Group, Department of Biochemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2H7.
Austin, J., and Kimble, J. (1987). glp-1 is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell 51, 589–599. Abstract Article
Denver, D.R., Morris, K., Lynch, M., Vassilieva, L.L., and Thomas, W.K. (2000). High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science 289, 2342–2344. Abstract Article
Dillin, A., Crawford, D.K., and Kenyon, C. (2002). Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 298, 830–834. Abstract Article
Dillin, A., Hsu, A.L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A.G., Kamath, R.S., Ahringer, J., and Kenyon, C. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. Abstract Article
Ewbank, J.J., Barnes, T.M., Lakowski, B., Lussier, M., Bussey, H., and Hekimi, S. (1997). Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275, 980–983. Abstract Article
Feng, J., Bussière, F., and Hekimi, S. (2001). Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644. Abstract Article
Garesse, R., and Vallejo, C.G. (2001). Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene 263, 1–16. Abstract Article
Garrido, N., Griparic, L., Jokitalo, E., Wartiovaara, J., van der Bliek, A.M., and Spelbrink, J.N. (2003). Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 14, 1583–1596. Abstract Article
González-Halphen, D., Funes, S., Pèrez-Martínez, X., Reyes-Prieto, A., Claros, M.G., Davidson, E., and King, M.P. (2004). Genetic correction of mitochondrial diseases: using the natural migration of mitochondrial genes to the nucleus in chlorophyte algae as a model system. Ann. N.Y. Acad. Sci. 1019, 232–239. Abstract Article
Gorbunova, V., and Seluanov, A. (2002). CLK-1 protein has DNA binding activity specific to OL region of mitochondrial DNA. FEBS Lett. 516, 279–284. Abstract Article
Gray, M.W., Burger, G., and Lang, B.F. (2001). The origin and early evolution of mitochondria. Genome Biol. 2, REVIEWS1018.1011-1018.1015. Abstract Article
Hartman, P., Ponder, R., Lo, H.H., and Ishii, N. (2004). Mitochondrial oxidative stress can lead to nuclear hypermutability. Mech. Ageing Dev. 125, 417–420. Abstract Article
Hartman, P.S., Ishii, N., Kayser, E., Morgan, P.G., and Sedensky, M.M. (2001). Mitochondrial mutations differentially affect aging, mutability and anesthetic sensitivity in Caenorhabditis elegans. Mech. Ageing Dev. 122, 1187–1201. Abstract Article
Hedgecock, E.M., and Thomson, J.N. (1982). A gene required for nuclear and mitochondrial attachment in the nematode Caenorhabditis elegans. Cell 30, 321–330. Abstract Article
Hihi, A.K., Gao, Y., and Hekimi, S. (2002). Ubiquinone is necessary for Caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J. Biol. Chem. 277, 2202–2206. Abstract Article
Ishii, N., Fujii, M., Hartman, P.S., Tsuda, M., Yasuda, K., Senoo-Matsuda, N., Yanase, S., Ayusawa, D., and Suzuki, K. (1998). A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394, 694–697. Abstract Article
Ishii, N., Goto, S., and Hartman, P.S. (2002). Protein oxidation during aging of the nematode Caenorhabditis elegans. Free Radic. Biol. Med. 33, 1021–1025. Abstract Article
Jacobs, H.T., Lehtinen, S.K., and Spelbrink, J.N. (2000). No sex please, we're mitochondria: a hypothesis on the somatic unit of inheritance of mammalian mtDNA. Bioessays 22, 564–572. Abstract
Jonassen, T., Larsen, P.L., and Clarke, C.F. (2001). A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc. Natl. Acad. Sci. USA 98, 421–426. Abstract Article
Jonassen, T., Marbois, B.N., Faull, K.F., Clarke, C.F., and Larsen, P.L. (2002). Development and fertility in Caenorhabditis elegans clk-1 mutants depend upon transport of dietary coenzyme Q8 to mitochondria. J. Biol. Chem. 277, 45020–45027. Abstract Article
Labrousse, A.M., Zappaterra, M.D., Rube, D.A., and van der Bliek, A.M. (1999). C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell 4, 815–826. Abstract Article
Lee, S.S., Lee, R.Y., Fraser, A.G., Kamath, R.S., Ahringer, J., and Ruvkun, G. (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48. Abstract Article
Lemieux, J., Lakowski, B., Webb, A., Meng, Y., Ubach, A., Bussière, F., Barnes, T., and Hekimi, S. (2001). Regulation of physiological rates in Caenorhabditis elegans by a tRNA-modifying enzyme in the mitochondria. Genetics 159, 147–157. Abstract
Lenaz, G., Baracca, A., Carelli, V., D'Aurelio, M., Sgarbi, G., and Solaini, G. (2004). Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim. Biophys. Acta 1658, 89–94. Abstract Article
Martin, A.M., Hammond, E., Nolan, D., Pace, C., Den Boer, M., Taylor, L., Moore, H., Martinez, O.P., Christiansen, F.T., and Mallal, S. (2003). Accumulation of mitochondrial DNA mutations in human immunodeficiency virus-infected patients treated with nucleoside-analogue reverse-transcriptase inhibitors. Am. J. Hum. Genet. 72, 549–560. Abstract Article
Melov, S., Hertz, G.Z., Stormo, G.D., and Johnson, T.E. (1994). Detection of deletions in the mitochondrial genome of Caenorhabditis elegans. Nucleic Acids Res. 22, 1075–1078. Abstract
Melov, S., Lithgow, G.J., Fischer, D.R., Tedesco, P.M., and Johnson, T.E. (1995). Increased frequency of deletions in the mitochondrial genome with age of Caenorhabditis elegans. Nucleic Acids Res. 23, 1419–1425. Abstract
Moraes, C.T. (2001). What regulates mitochondrial DNA copy number in animal cells? Trends Genet. 17, 199–205. Abstract Article
Okimoto, R., Macfarlane, J.L., Clary, D.O., and Wolstenholme, D.R. (1992). The mitochondrial genomes of two nematodes Caenorhabditis elegans and Ascaris suum. Genetics 130, 471–498. Abstract
Okimoto, R., Macfarlane, J.L., and Wolstenholme, D.R. (1990). Evidence for the frequent use of TTG as the translation initiation codon of mitochondrial protein genes in the nematodes Ascaris suum and Caenorhabditis elegans. Nucleic Acids Res. 18, 6113–6118. Abstract
Rube, D.A., and van der Bliek, A.M. (2004). Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem., 256-257, 331-339. Abstract Article
Schultz, B.E., and Chan, S.I. (2001). Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu. Rev. Biophys. Biomol. Struct. 30, 23–65. Abstract Article
Schwartz, M., and Vissing, J. (2002). Paternal inheritance of mitochondrial DNA. N. Engl. J. Med. 347, 576–580. Abstract Article
Senoo-Matsuda, N., Yasuda, K., Tsuda, M., Ohkubo, T., Yoshimura, S., Nakazawa, H., Hartman, P.S., and Ishii, N. (2001). A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J. Biol. Chem. 276, 41553–41558. Abstract Article
Starr, D.A., and Han, M. (2002). Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science 298, 406–409. Abstract Article
Timmis, J.N., Ayliffe, M.A., Huang, C.Y., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nature Rev. Genet. 5, 123–135. Abstract Article
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J.N., Rovio, A.T., Bruder, C.E., Bohlooly, Y.M., Gidlöf, S., Oldfors, A., Wibom, R., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. Abstract Article
Tsang, W.Y., and Lemire, B.D. (2002). Mitochondrial genome content is regulated during nematode development. Biochem. Biophys. Res. Commun. 291, 8–16. Abstract Article
Tsang, W.Y., and Lemire, B.D. (2002). Stable heteroplasmy but differential inheritance of a large mitochondrial DNA deletion in nematodes. Biochem. Cell Biol. 80, 645–654. Abstract
Tsang, W.Y., and Lemire, B.D. (2003). The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochim. Biophys. Acta 1638, 91–105. Abstract
Tsang, W.Y., Sayles, L.C., Grad, L.I., Pilgrim, D.B., and Lemire, B.D. (2001). Mitochondrial respiratory chain deficiency in Caenorhabditis elegans results in developmental arrest and increased lifespan. J. Biol. Chem. 276, 32240–32246. Abstract Article
Van Voorhies, W.A., and Ward, S. (1999). Genetic and environmental conditions that increase longevity in Caenorhabditis elegans decrease metabolic rate. Proc. Natl. Acad. Sci. USA 96, 11399–11403. Abstract Article
Wolstenholme, D.R. (1992). Animal mitochondrial DNA: structure and evolution. In Mitochondrial Genomes. Wolstenholme, D.R., and Jeon, K.W. eds., (San Diego, California: Academic Press, INC.), Vol. 141, pp. 173–216. Abstract
*Edited by Jonathan Hodgkin and Philip Anderson. Last revised January 4, 2005. Published September 14, 2005. This chapter should be cited as: Lemire, B. Mitochondrial genetics (September 14, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.25.1, http://www.wormbook.org.
Copyright: © 2005 Bernard Lemire. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: bernard.lemire@ualberta.ca
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.