Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
The architecture and dynamics of molecular networks can provide an understanding of complex biological processes complementary to that obtained from the in-depth study of single genes and proteins. With a completely sequenced and well-annotated genome, a fully characterized cell lineage, and powerful tools available to dissect development, Caenorhabditis elegans, among metazoans, provides an optimal system to bridge cellular and organismal biology with the global properties of macromolecular networks. This chapter considers omic technologies available for C. elegans to describe molecular networks - encompassing transcriptional and phenotypic profiling as well as physical interaction mapping - and discusses how their individual and integrated applications are paving the way for a network-level understanding of C. elegans biology.
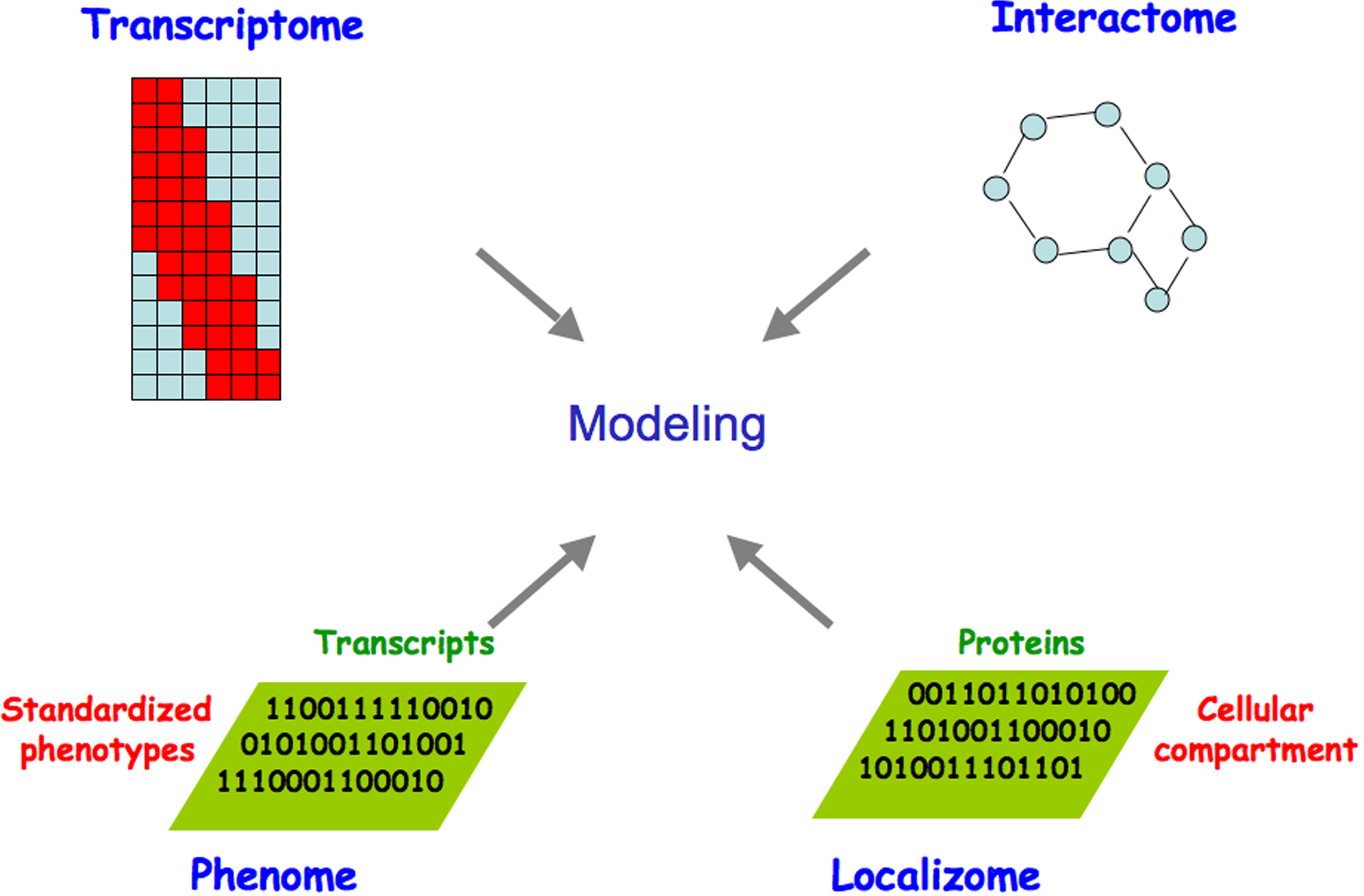
The combined power of global approaches to systematically analyze interactions among genes and their products and the phenotypic consequences of in vivo perturbations promises to drive the development of increasingly sophisticated models of the topology, function, and dynamics of C. elegans molecular networks (Figure 1). The C. elegans genome sequence and its annotation laid the foundation for the development of a variety of functional genomic or “omic” approaches to interrogate molecular networks. This chapter reviews the developments underpinning these technologies, giving particular attention to: i) identifying network components, i.e. the complete lists of non-coding and coding transcripts and open reading frames (ORFs); ii) mapping interconnections between these components; iii) systematic perturbation of networks; iv) spatiotemporal dynamics of network components; and v) global network modeling. We summarize the resources emerging from these efforts, current experimental and computational approaches including the integration of different omic datasets, and developments on the horizon.
|
Figure 1. Omic approaches to generate gradually improving models of the complete set of biologically relevant interactions. Genome- and proteome-scale studies have provided information on the topology, function and dynamics of C. elegans macromolecular networks. Expression profiling (transcriptome), DNA-protein, RNA-protein, and protein-protein interactions (interactome), phenotypic analyses (phenome), and protein localization (localizome) can be integrated, allowing the dynamic properties of the resulting network to be related to C. elegans biology. Adapted from Walhout et al. (1998) and Vidal (2001).
The initial release of the assembled, annotated C. elegans genome sequence (The C. elegans Sequencing Consortium, 1998) was a milestone in genomics, marking the first sequenced genome from a multicellular organism. Importantly for comprehensive analyses, C. elegans remains the only multicellular organism for which a complete genome sequence is available with no gaps in the assembly (Hillier et al., 2005).
An innovative object-oriented database system, ACeDB (Durbin and Thierry-Mieg, 1994; Stein et al., 2001; see also http://www.acedb.org/Cornell/dkfz.html), was created to facilitate the C. elegans genome project. ACeDB was the first widely used database system designed specifically for capturing and archiving complex biological data. Coupling the database to a visual output anchored on the genomic sequence gave ACeDB a graphical user interface, allowing the C. elegans community to extract far more information than was previously possible. From ACeDB evolved WormBase, a collaborative effort to capture, curate, and distribute biological information on C. elegans and related nematodes (Stein et al., 2001; Chen et al., 2005), and the WormGenes database at NCBI, which also provides access to carefully curated C. elegans genome annotations and functional data (Thierry-Mieg et al., 2005). These databases are invaluable sources of information for the C. elegans research community (see Table 1 for a short guide to these and other useful online resources for C. elegans).
Table 1. Web resources for C. elegans.
| Website | URL | Information provided |
|---|---|---|
| WormBase | http://www.wormbase.org | C. elegans genome annotation, including datasets and reagents for YK cDNAs, ORFeome clones, RNAi results, microarray experiments, Y2H and genetic interactions, and much more |
| WormGenes | http://www.wormgenes.org | C. elegans genome annotation with carefully curated YK cDNA models and literature data |
| Intronerator | http://www.soe.ucsc.edu/~kent/intronerator | Tools for exploring the molecular biology and genomics of C. elegans with a special emphasis on alternative splicing |
| C. elegans WWW server | http://elegans.swmed.edu | Leon Avery's links to all things nematode |
| WORFDB | http://worfdb.dfci.harvard.edu | ORFeome clones and associated data |
| NextDB | http://nematode.lab.nig.ac.jp/dbest/keysrch.html | cDNA library clones and associated data |
| http://nematode.lab.nig.ac.jp/db2/index.php | In situ mRNA expression patterns | |
| Hope Lab Expression Pattern Database | http://129.11.204.59:591/fmi/iwp/cgi?-db=Construct%20Info&-loadframes | Promoter::GFP fusion expression patterns |
| BC Gene Expression Consortium | http://elegans.bcgsc.ca/home/ge_consortium.html | SAGE data |
| Promoter::GFP expression patterns | ||
| WormAtlas | http://www.wormatlas.org | Behavioral and structural anatomy |
| WormImage | http://www.wormimage.org | Electron micrographs and associated data |
| C. elegans Gene Knockout Consortium | http://celeganskoconsortium.omrf.org | Gene knockout strains |
| RNAiDB | http://www.rnai.org | RNAi phenotypes and associated data for most publicly available RNAi studies |
| PhenoBank | http://www.worm.mpi-cbg.de/phenobank2/cgi-bin/MenuPage.py | RNAi phenotypes and associated data from Sönnichsen et al. (2005) |
| http://www.phenobank.org | ||
| Predictions of C. elegans Genetic Interactions | http://tenaya.caltech.edu:8000/predict | Zhong and Sternberg (2006) |
| NBrowse | http://www.gnetbrowse.org | Integrated functional network browser |
At the time of the initial genome sequence release, GeneFinder (Green and Hillier, unpublished software) was used to predict approximately 20,000 protein coding sequences (CDSs) or ORFs. Alternative ab initio transcript or ORF prediction algorithms produce largely consistent results that differ primarily in predicted exon/intron junctions (Stein et al., 2003; Wei et al., 2005). Experimental evidence (see below) indicates that most of the ~20,000 predicted ORFs are at least transcribed (Reboul et al., 2001; Reboul et al., 2003). Approximately 1,300 noncoding RNA (ncRNA) transcripts have also been predicted, including at least 120 microRNAs (miRNAs) (see C. elegans noncoding RNA genes).
Accurate genome annotation requires both a high quality genome sequence and robust computational prediction tools. However, experimental validation of transcribed mRNA sequences and their protein products is indispensable to demonstrate transcription and splicing and to ensure that exon/intron boundaries and reading frames are correctly assigned. These companion activities together lead to the refinement of gene models through an iterative process. After an initial effort by the C. elegans Sequencing Consortium to sequence random expressed sequence tags (ESTs) (Waterston et al., 1992), a long-term effort was dedicated to isolating, sequencing and distributing as many complementary DNA (cDNA) clones as possible, with the ultimate goal of obtaining full-length cDNAs for all protein-coding transcripts (see Table 1). This benefited the worm community by providing cDNAs that helped refine transcript models and enabled cloning experiments performed in many laboratories over the years. Data from this cDNA collection are available from the National Institute of Genetics in Japan (Table 1) and through WormGenes and WormBase, including details on sequences and corresponding gene products. The latest WormBase frozen release (WS150) contains over 250,000 EST reads from this library, matching ~10,500 distinct genes. Around 2,000 of these cDNAs are likely full-length clones. WormBase, WormGenes, and Intronerator (Kent and Zahler, 2000) all offer graphical representations of the collection that make it very convenient to inspect the precise structure of transcript models along with all supporting evidence (Table 1).
Because most transcripts are expressed at relatively low levels, random sequencing of clones from cDNA libraries makes it extremely hard to capture an entire set of full-length cDNAs (Reboul et al., 2003), and the rate of novel sequences identified has reached diminishing returns due to the relative abundance of encoded transcripts in the library (Das et al., 2001). This is true not only for C. elegans but also for other organisms (Carninci et al., 2003; Castelli et al., 2004). In addition, although full-length cDNAs provide crucial information to annotate transcripts, their 5′ and 3′ UTR sequences usually prevent their use for large-scale protein expression studies. Finally, the vectors used in cDNA cloning projects are usually not designed for automated cloning and convenient proteome-wide protein expression (Brasch et al., 2004). For these reasons, a C. elegans “ORFeome project” was initiated with the goal of generating Gateway recombinational clones for nearly all ORFs (i.e. precisely from the predicted initiation to the predicted termination codons) (Walhout et al., 2000; Reboul et al., 2001; Reboul et al., 2003; Lamesch et al., 2004).
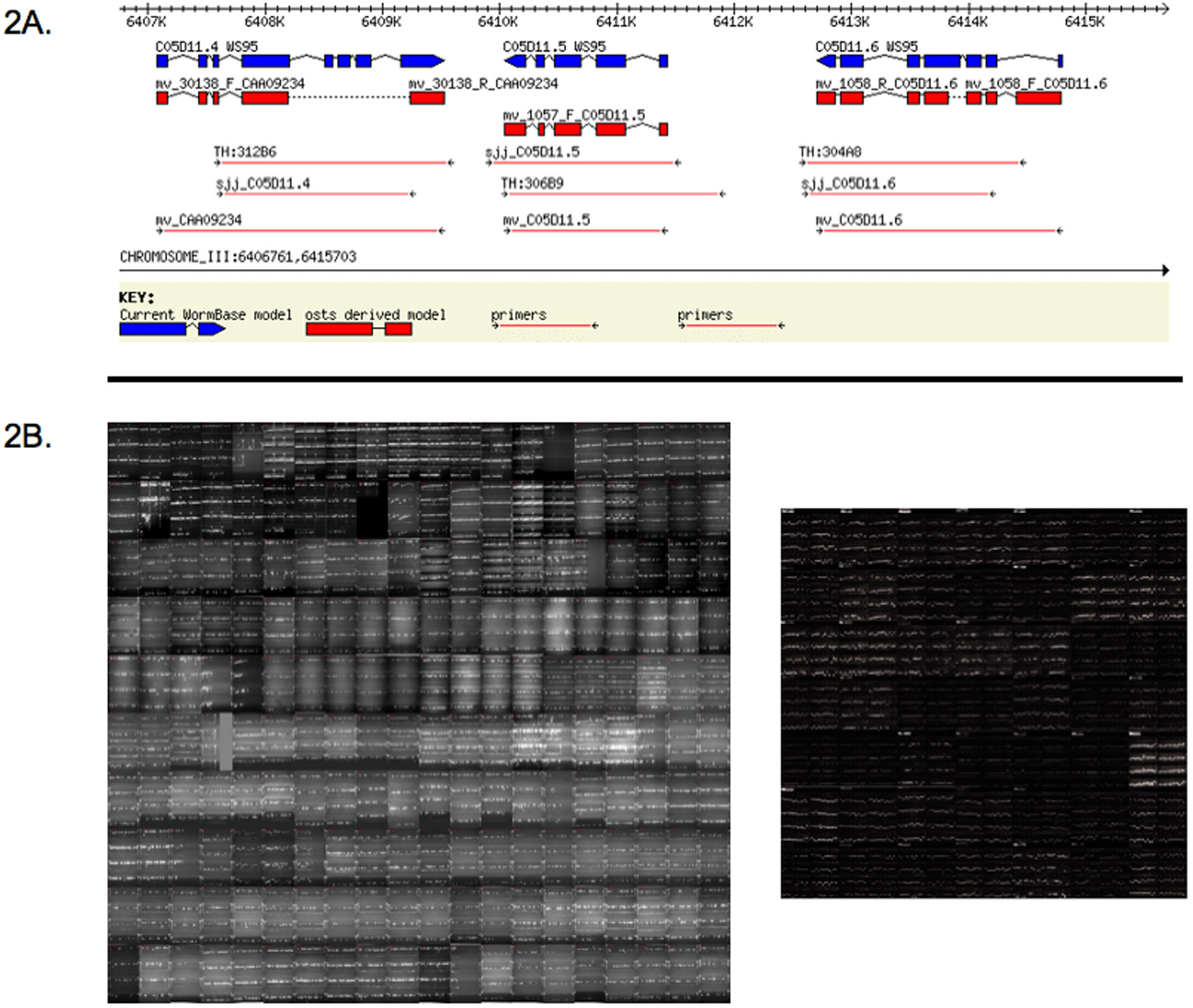
Rather than picking and sequencing individual clones from cDNA libraries, PCR amplification carried out with primer pairs specific to ORFs and with a cDNA library or reverse-transcribed polyA+ mRNA as template can achieve nearly complete coverage (Reboul et al., 2001). Using the ~20,000 ORF predictions from WS9 (released in 1999), nearly 11,000 ORF constructs suitable for protein expression were initially obtained (Reboul et al., 2003) (Figure 2). Over 4,000 of these had no prior experimental support, demonstrating that ab initio ORF predictions could indeed be verified experimentally. The nearly 8,000 unsuccessful attempts were due to imprecise exon/intron predictions, usually surrounding the putative translation start site (Reboul et al., 2001). From 1999 to 2003, updates to WormBase led to over 4,200 new or revised ORF predictions, based on a combination of improved algorithms, comparative genomics, and experimental evidence from cDNA and ORF sequences. Of these repredicted ORFs, ~2,500 were cloned in Version 3.1 of the C. elegans ORFeome project (Lamesch et al., 2004).
|
Figure 2. The C. elegans ORFeome. A) Schematic representation of predicted exon/intron structures for three C. elegans ORFs from WS9 of WormBase. Blue boxes correspond to predicted exons based on Genefinder. Based on ORF sequence tags (OSTs) obtained from C. elegans cloned ORFs are aligned to the genome (red boxes), the structures of two ORFs match the Genefinder prediction whereas one differs from prediction. B) Agarose gels showing PCR products for ORFeome clones. The ~10,000 ORFs in C. elegans ORFeome Version 1.1 (Reboul et al., 2003) (left-hand panel, 200 gels) were supplemented by an additional ~2,500 ORFs in Version 3.1 (right-hand panel, 49 gels) based on improved annotations in WormBase WS100 (Lamesch et al., 2004).
In total, there is currently experimental evidence for ~18,000 protein-coding ORFs. WormBase release WS150 contains over 22,000 ORF entries, of which ~2,800 are confirmed isoforms from alternative splicing, and ~4,800 are predicted ORFs with no experimental validation. The majority of clones from the ORFeome and cDNA collections have been end-sequenced (generating ESTs and OSTs) but not yet fully characterized. Sequencing of several hundred ORF clones suggests that more isoforms exist than are currently annotated in WormBase, and that the number of genes with alternative splicing is probably in the range of 15%, (Reboul et al., 2003). A complete and accurate inventory of all isoforms represented by fully sequenced, wild-type clones awaits further experimental validation guided by gene prediction algorithms - such as Twinscan (e.g. Twinscan Korf et al., 2001; Wei et al., 2005) and others - that can take advantage of sequence comparisons with other Caenorhabditis species (Stein et al., 2003; see The phylogenetic relationships of Caenorhabditis and other rhabditids; NHGRI, 2005). Access to precise gene structural information has enabled the development of multiple genome-scale resources that can be used for a variety of applications (Table 2), transforming the way experimental science is conducted. Overall, one important lesson learned from the C. elegans cDNA and ORFeome projects is that genome annotation and experimental verification are complementary activities that advance understanding through an iterative process.
Table 2. Collections of reagents for functional genomics and proteomics in C. elegans.
| Organization | URL or contact | Resource |
|---|---|---|
| Sanger Institute | Send inquires to Audrey Fraser (aef@sanger.ac.uk) | Genomic (YAC and cosmid) clones (Currently Unavailable) |
| Kohara lab | Send requests to Yuji Kohara (ykohara@lab.nig.ac.jp) | Kohara cDNA library clones |
| GeneService LTD | http://www.geneservice.co.uk/products/rnai/ | Ahringer lab RNAi feeding library (Kamath et al., 2003) |
| http://www.geneservice.co.uk/products/clones/ Celegans_Fos.jsp | BC Fosmid library (D. Moerman et al., unpublished) | |
| Open Biosystems | http://www.openbiosystems.com/Genomics/ Model%20Organism%20Resources/ Worm%20Resources/ | ORFeome library (Reboul et al., 2003; Lamesch et al., 2004) |
| ORFeome-RNAi feeding library (Rual et al., 2004) | ||
| Promoterome library (Dupuy et al., 2004) | ||
| Wash U Genome Sequencing Center | http://www.genome.wustl.edu/genome/ celegans/microarray/ma_gen_info.cgi | 70-mer oligonucleotide microarrays (Washington University Genome Sequencing Center, 2005) |
| Affymetrix | http://www.affymetrix.com/products/ arrays/specific/celegans.affx | C. elegans GeneChip genome arrays |
| Caenorhabditis Genetics Center (CGC) | http://www.cbs.umn.edu/CGC/ | C. elegans strains (including gene knockout strains from the C. elegans Gene Knockout Consortium and some GFP transgenic strains from the BC Gene Expression Consortium) |
| BC Gene Expression Consortium | http://elegans.bcgsc.ca/perl/eprofile/contact | Promoter::GFP transgenic strains (McKay et al., 2003) |
Proteins do not function in isolation, but rather as interacting partners in complexes (Alberts, 1998), modules and networks (Hartwell et al., 1999). Thus a major challenge is to establish the full set of all protein-protein, DNA-protein and RNA-protein interactions, or to map the “interactome” network. Binary protein-protein interactions can be readily identified using the yeast two-hybrid (Y2H) system (Fields and Song, 1989; Vidal and Legrain, 1999), with the advantage that this system is amenable to high-throughput proteome-wide mapping projects (Walhout and Vidal, 2001). When generated with appropriate caution and necessary controls, Y2H datasets can be of high quality (Vidalain et al., 2004).
The C. elegans interactome mapping project was initiated in the context of specific biological processes such as vulval development, protein degradation, germline development, DNA damage response and Dauer formation (Walhout et al., 2000; Davy et al., 2001; Boulton et al., 2002; Walhout et al., 2002; Tewari et al., 2004). One of the main lessons drawn by overlapping these datasets obtained up to that point was a much higher level of molecular connectivity between seemingly different pathways than previously imagined (Reboul et al., 2003). The project then moved to a “first-draft” proteome-scale C. elegans interactome map primarily focused on metazoan-specific proteins (Li et al., 2004). The current version of the global C. elegans Y2H-based interactome network contains ~5,500 potential interactions, comprising approximately 10% of the expected number of protein-protein interactions (Li et al., 2004). The validation rate of the interactome map was estimated at ~70% by retesting large numbers of interactions using a completely different interaction assay (Li et al., 2004), suggesting a high level of quality overall.
Like other interactome networks, the topology of the C. elegans interactome appears “scale-free” (a few proteins have many interacting partners whereas most proteins interact with only a few partners) and “small-world” (any two proteins can be connected through a chain of only a few interactions) (Barabasi and Albert, 1999; Goldberg and Roth, 2003; Li et al., 2004). These and other topological properties of global interactome networks might relate to biological properties such as robustness and plasticity (Jeong et al., 2001; Han et al., 2004).
Completing the interactome map will require both a nearly complete C. elegans ORFeome collection as well as additional approaches that utilize modified versions of either Y2H or affinity-purification of protein complexes, since no single method for capturing protein-protein interactions is 100% effective (Walhout et al., 2000). As with efforts to accurately annotate the genome, interactome mapping projects will necessarily proceed iteratively through the interplay of direct experimentation and computational efforts (Vidal, 2005).
While physical interaction maps help elucidate molecular events at the biochemical level, other functional assays can reveal logical connections between gene products that are required for the same set of biological processes. RNAi (Guo and Kemphues, 1995; Fire et al., 1998) is amenable to medium to high throughput approaches that can be used to systematically knock down large numbers of transcripts, thereby relating perturbations of molecular networks to phenotypic analyses and setting the stage for generating a global phenotypic or “phenome” map of C. elegans (reviewed in Gunsalus and Piano, 2005). The foundation for a phenome map of early embryogenesis has been laid by a host of recent large-scale RNAi studies, which have provided at least a first-pass phenotypic analysis for nearly every protein-coding gene in the genome (Fraser et al., 2000; Gönczy et al., 2000; Piano et al., 2000; Maeda et al., 2001; Piano et al., 2002; Kamath et al., 2003; Simmer et al., 2003; Rual et al., 2004; Fernandez et al., 2005; Sönnichsen et al., 2005). Results from various RNAi studies in C. elegans are available online through RNAiDB (Gunsalus et al., 2004), PhenoBank (Sönnichsen et al., 2005), and WormBase (see Table 1).
Questions of reliability and completeness naturally arise regarding genome-scale RNAi analyses. Comparisons with genetic analyses have found that around 25% of the loci for which genetic alleles confer embryonic phenotypes are typically not recovered in large-scale RNAi studies in the laboratory strain N2 (Kamath et al., 2003; Fernandez et al., 2005; Sönnichsen et al., 2005). In contrast, comparisons of embryonic lethality between genetic and high confidence RNAi results indicate a very low level (generally <1%) of false positives (Kamath et al., 2003; Simmer et al., 2003; Rual et al., 2004; Fernandez et al., 2005). Potential false positive results could arise from the depletion of multiple transcripts by a single RNAi construct that inadvertantly targets multiple genes (Gunsalus et al., 2004; Qiu et al., 2005). The actual off-target error rate in vivo will depend on factors such as cooperativity between short interfering RNAs (siRNAs), mRNA abundances in different tissues, and saturation of the RNAi machinery. RNAiDB and WormBase both provide data on potential off-target effects for RNAi reagents used by the worm community, calculated using different methods.
It is interesting to note that the proportion of essential genes appears to be in a similar range (~15–25%) in yeast (Winzeler et al., 1999; Giaever et al., 2002), worms (see Essential genes), and flies (Ashburner et al., 1999). This apparent similarity across phyla may reflect a universal property of networks, arising from a need to evolve network connectivity in order to maintain a balance between robustness (to survive perturbations) and plasticity (to explore new phenotypic space). An observation consistent with this idea is that the penetrance level observed in the RNAi analysis of essential genes is proportional to how ancient the targeted protein is (Fernandez et al., 2005).
Global RNAi studies are valuable not only to generate hypotheses for individual proteins, analogous to conventional genetic screening strategies, but also to address higher-order questions about groups of functionally related proteins. A set of defined phenotypes resulting from perturbations can be systematically scored for both positive and negative effects, defining “phenotypic profiles”. Gene products can then be grouped into “phenoclusters” based on similarity between phenotypic profiles (Vidal, 2001; Piano et al., 2002). For example, two distinct phenoclusters were found among two dozen proteins implicated in a DNA damage response interactome map (Boulton et al., 2002). One phenocluster predicted novel DNA repair proteins, while the other suggested novel proteins involved in the DNA damage induced checkpoint response.
In a set of comprehensive studies, RNAi phenotypes in the early embryo were analyzed using a systematic approach to explicitly score over 40 specific cell biological features (Piano et al., 2002; Sönnichsen et al., 2005). The resulting “phenotypic signatures” were used to cluster genes into groups of similar function, such as chromosome segregation or nuclear import-export. In a single study analyzing RNAi phenotypes for 98% of C. elegans genes, nearly all of the 661 genes identified genome-wide to elicit an RNAi phenotype in the early embryo could be placed into 23 separable phenotypic classes (Sönnichsen et al., 2005). In addition numerous hypotheses could be formulated as to how molecular networks are coordinated to drive early embryogenesis (Gunsalus et al., 2005).
The development of automated image analysis could alleviate what is currently a principal bottleneck in phenotypic analysis: the manual scoring of phenotypes by visual inspection. Recently a formal classification system linked to an automated image analysis system was developed that is capable of automatically gathering many behavioral and gross morphological phenotypic parameters (Geng et al., 2003; Feng et al., 2004; Geng et al., 2004). This system has been used to classify genotypes based on automatically extracted image features. Automated image analysis pipelines are also under development for analyzing cell lineage patterns (Yasuda et al., 1999; Bao et al., 2006) and high-content embryonic phenotypes (Ning et al., 2005).
The transcriptome and proteome at any given time and place constitute only a subset of all possible macromolecules potentially encoded in the genome: different combinations of expressed gene products are present in different cells at different times, move within and between cellular compartments, and interact with a variety of other molecules to carry out their functions. Thus to relate C. elegans cellular and organismal biology to global properties of molecular networks, it will become increasingly important to establish when and where the complete repertoire of coding and noncoding transcripts and protein products are expressed and localized throughout development, both at a cellular and sub-cellular level. Such a “localizome” map would be extremely valuable in the context of C. elegans since a nearly perfect anatomical atlas has already been generated, providing a complete description of the cell lineage and a map of the neuronal network (Sulston and Horvitz, 1977; White et al., 1982; Sulston et al., 1983).
Technological innovations to parallelize studies of gene expression patterns have enabled genome-scale analyses of increasing sophistication to discover the spatiotemporal activity of promoters, to define sets of co-regulated genes, to identify shared cis-acting regulatory sites and corresponding trans-acting factors, and to use these data to build models of transcriptional networks and regulatory control mechanisms. Gene expression in C. elegans is being evaluated using a variety of methods, including microarrays (Hill et al., 2000; Reinke et al., 2000), serial analysis of gene expression (SAGE) (Jones et al., 2001; Pleasance et al., 2003), quantitative reverse-transcription with PCR (Q-RT-PCR) (reviewed in Bustin, 2000), and in situ analyses of mRNA expression patterns (Table 2). Tiling arrays - now in use for human (Bertone et al., 2004), Drosophila (Stolc et al., 2004), and Arabidopsis (Yamada et al., 2003) - should soon reveal alternative splicing patterns and expressed non-coding regions of the genome, providing further experimental data for improving genome annotation and defining the repertoire of functional genetic elements. See Tables 1-3 for representative examples of reagents and datasets.
Specific developmental processes, temporal transitions, or environmental responses in C. elegans have been examined by measuring gene expression in wild type and genetically altered, RNAi-treated, or environmentally challenged animals using microarray and SAGE (Table 3 provides a sampling of these studies). Remarkably, it has been possible to isolate mRNA from single tissues or cell types such as muscle or neurons, using highly specific promoters to drive transgenic constructs expressing either poly-A binding protein followed by co-immunoprecipitation (co-IP) (Roy et al., 2002; Kunitomo et al., 2005) or a fluorescent marker followed by fluorescence activated cell sorting (FACS) (Zhang et al., 2002; Colosimo et al., 2004; Cinar et al., 2005; Fox et al., 2005).
Table 3. A sampling of dynamic transcriptome analyses of developmental processes or environmental responses in C. elegans.
| Biological focus | Reference |
|---|---|
| Developmental transitions | Hill et al. (2000); Jiang et al. (2001); Baugh et al. (2003); Robertson et al. (2004) |
| Germline development | Reinke et al. (2000); Reinke et al. (2004) |
| Meiosis | Colaiacovo et al. (2002) |
| Lineage specification | Baugh et al. (2005) |
| Ethanol response | Kwon et al. (2004) |
| Radiation response | Nelson et al. (2002) |
| Heat shock response | GuhaThakurta et al. (2002) |
| Developmental roles of fatty acids | Kniazeva et al. (2004) |
| Translation initiation factors | Dinkova et al. (2005) |
| Amyloid beta-responsive genes | Link et al. (2003) |
| Aging / longevity | Lund et al. (2002); McCarroll et al. (2004) |
| Dauer pathway | Holt and Riddle (2003); Wang and Kim (2003); McElwee et al. (2004) |
| Hypertonic stress resistance | Lamitina and Strange, 2005) |
| Muscle-specific genes | Roy et al. (2002) |
| Neuronal genes | Zhang et al. (2002); Colosimo et al. (2004); Cinar et al. (2005); Fox et al. (2005); Kunitomo et al. (2005). |
To identify gene regulatory elements controlling expression and the logical circuitry driving transcriptional networks, both computational and experimental approaches are being pursued. Informatic analyses have provided insights into co-regulated groups of genes, either within C. elegans (Kim et al., 2001; Owen et al., 2003), or conserved across species (Stuart et al., 2003; McCarroll et al., 2004). Predictions of cis-regulatory elements based on conserved sequence motifs, either among co-expressed genes (GuhaThakurta et al., 2002; Zhang et al., 2002; Kwon et al., 2004; Cinar et al., 2005) or gene families (McCarroll et al., 2005), or in combination with positional and combinatorial constraints (Beer and Tavazoie, 2004), are generating testable hypotheses on gene regulatory circuitry and rules. Experimental efforts to decipher network logic are employing techniques such as yeast one-hybrid assays (Deplancke et al., 2004; Deplancke et al., 2006), in vivo analysis of green fluorescent protein (GFP) reporter expression (Chalfie et al., 1994), and chromatin immunoprecipitation (ChIP-chip technology; Hanlon and Lieb, 2004; Oh et al., 2006). The recent demonstration that functional GFP can be reconstituted from fragments driven by separate promoters (Zhang et al., 2004) opens the gate to large-scale systematic analysis to delineate combinatorial control in vivo.
Increasingly, combined experimental and informatic efforts seek to characterize higher-order features of transcriptional networks. These global approaches have discovered, for example, the chromosomal clustering of muscle-expressed genes (Roy et al., 2002), or different classes of radiation-modulated genes (Nelson et al., 2002). Finally, comparative studies are addressing questions such as the evolution of gene expression patterns (Denver et al., 2005) and cis-regulatory elements (Castillo-Davis et al., 2004). While these studies represent just an early foray into the analysis of gene regulatory networks, significant strides in this area can be anticipated in the near future.
High-resolution data on spatiotemporal expression patterns throughout development can be obtained using microscopic techniques. A large-scale in situ analysis of mRNA is being carried out using cDNAs (see Table 1) to identify when and where transcripts are expressed in the worm. In addition, C. elegans provides an unparalleled opportunity to map dynamic patterns of expression in vivo, due to its invariant and comprehensively identified cell lineage (Sulston and Horvitz, 1977; White et al., 1982; Sulston et al., 1983) and the fact that animals are optically transparent throughout the life cycle. Transgenic animals containing different tissue-specific promoters driving GFP provide surrogate markers for expression, and protein-GFP fusion reporter constructs allow the direct visualization of subcellular localization. Systematic in vivo analyses of expression patterns are being carried out for thousands of genes (see Table 1) (McKay et al., 2003; Hope et al., 2004; Zhao et al., 2004; Baillie and Moerman, 2005), using transgenic worms that harbor reporter constructs containing GFP fused downstream of cloned intergenic promoter regions (Hope et al., 1996; Dupuy et al., 2004). This approach requires the ability to efficiently generate large numbers of transgenic worm strains transformed with cloned DNA fusion constructs and, ideally, should faithfully report promoter activity in vivo. The systematic large-scale application of this approach will require relatively convenient scoring by microscopy in live animals and will rely heavily on ongoing efforts to develop sophisticated imaging technologies (Mohler and White, 1998; Mohler et al., 2003; Bao et al., 2006). Collections of transgenic strains (e.g. carrying GFP reporter constructs) are being made available for distribution (Table 2).
The idea that meaningful hypotheses of gene function and network properties may be obtained by combining two or more types of functional associations (reviewed in Vidal, 2001; Ge et al., 2003) is being explored using a variety of approaches. Global evidence suggests that genes with similar expression profiles are more likely to encode interacting proteins (Ge et al., 2001; Grigoriev, 2001; Jansen et al., 2002; Kemmeren et al., 2002). Correlations between protein-protein interactions and phenotypic data have also been observed, both in yeast (Said et al., 2004) and among C. elegans DNA damage response (DDR) (Boulton et al., 2002) and germline-enriched genes (Walhout et al., 2002).
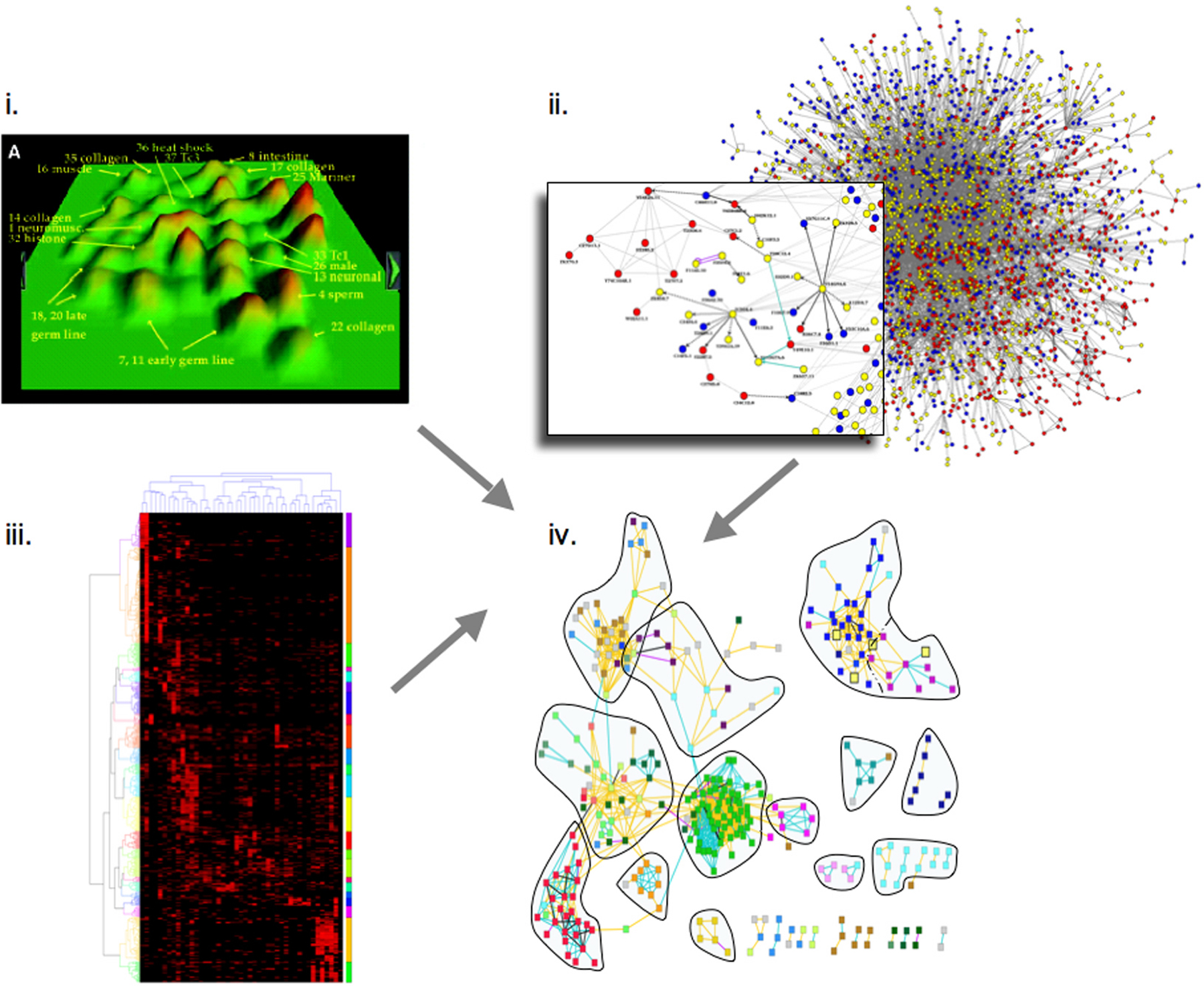
Extending a genome-wide phenotypic analysis of genes required in the early embryo, a combined analysis of high-content phenotypic data (Sönnichsen et al., 2005), transcriptional correlations (Kim et al., 2001), and protein-protein interaction data (Li et al., 2004) has revealed correlations between all three data types (Gunsalus et al., 2005) (Figure 3). It appears that on a global scale, highly interconnected proteins give rise to the same range of phenotypes upon depletion by RNAi, and quantitatively similar phenotypes are predictive of protein-protein interactions. Extending this logic across the proteins found to be required for early embryonic events revealed assemblies of functionally linked components, generating testable functional predictions for numerous unknown genes. This analysis suggests that a limited number of interconnected “molecular machines” drive early embryogenesis and provides a global blueprint to explore how protein complexes are coordinated (Gunsalus et al., 2005).
Understanding how functional interactions between genes affect biological processes will ultimately require combinatorial in vivo perturbations. Since experimentally testing the 200 million possible pairwise combinations between 20,000 genes for each type of experimental perturbation (e.g., reduced or increased level of activity, or alteration of specific contacts) is currently intractable, computational methods are needed that can integrate heterogeneous data and help focus experimental efforts on pairs that may be more likely to be functionally related. A recent study has made genome-wide predictions of genetic interactions in C. elegans using a logistic regression approach to integrate data on protein-protein interactions, gene expression, and phenotypes from yeast, worm, and fly with available functional annotations (Zhong and Sternberg, 2006). Like the early embryonic network described above, the resulting network of over 18,000 potential genetic interactions between about 10% of genes in C. elegans suggests a modular organization of protein complexes and specific cellular processes. These predictions provide a step toward a global view of genetic interactions in a multicellular organism.
Visualizing multidimensional maps is essential to navigate the vast amounts of data now accumulating. To make the results of integrated network analyses more easily accessible, a web-based interactive graphical tool called “N-Browse” has been implemented to allow browsing integrated networks of physical and logical functional connections between genes and their products in C. elegans and other species (Table 1).
|
Figure 3. Combined analysis of (i.) transcriptional correlations (Kim et al., 2001); reprinted with permission from Kim et al. (2001). Copyright 2001 American Association for the Advancement of Science. (ii.) protein-protein interaction data (Li et al., 2004), and (iii.) high-content phenotypic data (Sönnichsen et al., 2005). The three datasets are integrated (iv.) revealing correlations between all three data types (Gunsalus et al., 2005). Extending this logic across all protein-coding genes found to be required for early embryonic events reveals assemblies of functionally linked components, allowing testable functional predictions for numerous proteins.
The application of omic approaches to elucidate C. elegans network biology promises to extend our knowledge of cellular and organismal biology as more efforts are dedicated to the development of resources and the generation of improved interactome, phenome and localizome datasets. First, it will be important to improve and eventually complete genome-wide resources such as the ORFeome, the Promoterome, and other sets of important DNA sequences such as 5′ and 3′ untranslated regions (UTRs), miRNAs and other noncoding RNAs. Second, it is critical to continue improving the mapping of molecular networks by developing and implementing new protein-protein interaction assays (Vidal, 2005) and other types of methods to assay DNA-protein (Deplancke et al., 2004) and RNA-protein (SenGupta et al., 1996) interactions. Third, it will be increasingly important to expand the phenome map by increasing the number of discrete phenotypes scored systematically and by improving and extending flexible RNAi resources (Rual et al., 2004), phenotype scoring methodologies, and image analysis pipelines. Fourth, the localizome map is currently only in its infancy, and it will be interesting to see how its development will help bridge network biology with the nearly perfect anatomy atlas available for C. elegans. Fifth, because disruption of biologically relevant interactions may be expected to phenocopy either null or gain-of-function alleles, it will be useful to exploit strategies for generating and using mutations that specifically perturb individual interactions (Vidal et al., 1996; Vidal et al., 1996; Endoh et al., 2000; Endoh et al., 2002).
The combination of experimentation and informatic analysis in iterative cycles will take on a central role in the future of biological research. As more data from functional and proteomic mapping projects are generated and more sophisticated methods are applied to integrate these data on a genome/proteome scale, their synthesis should prove increasingly informative for the reconstruction of cellular networks and for generating new testable hypotheses of biological systems. The challenge is now to achieve a true synthesis of the data to obtain a system-level view of the underlying regulatory networks and the evolutionary forces that have shaped them. This will require the marriage of large datasets with computational models that can describe and predict the behavior of dynamic macromolecular networks and their relationship to the underlying biological processes they control.
The C. elegans ORFeome, interactome and localizome projects at the Center for Cancer Systems Biology (CCSB) at the Dana-Farber Cancer Institute are funded by a NHGRI/NIGMS grant awarded to MV and F. Roth and NCI grants awarded to MV. The phenome and network visualization projects at the Center for Comparative Functional Genomics (New York University) are funded by an NICHD grant to FP and support from the U.S. Army and NYSTAR to KCG. We thank D. Baillie, M. Brent, D. Fitch, I. Hope, J. Hubbard, D. Moerman, B. Mohler, N. Rajewsky, V. Reinke, F. Roth, D. Thierry-Mieg, J, Thierry-Mieg, S. Vandenheuvel and M. Walhout for productive and friendly collaborations, and M. Walhout and M. Cusick for critical reading of the manuscript. Finally, we thank the C. elegans Sequencing Consortium for having generated the complete genome sequence and provided a high quality genome annotation, and WormBase for ever-improving gene models.
Alberts, B. (1998). The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92, 291–294. Abstract Article
Ashburner, M., Misra, S., Roote, J., Lewis, S.E., Blazej, R., Davis, T., Doyle, C., Galle, R., George, R., Harris, N., et al. (1999). An exploration of the sequence of a 2.9-Mb region of the genome of Drosophila melanogaster: the Adh region. Genetics 153, 179–219. Abstract
Baillie, D.L., and Moerman, D. (2005). BC C. elegans Gene Expression Consortium. (http://elegans.bcgsc.ca/home/ge_consortium.html)
Bao, Z., Murray, J.I., Boyle, T., Ooi, S.L., Sandel, M.J., and Waterston, R.H. (2006). Automated cell lineage tracing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 103, 2707–2712. Abstract Article
Barabasi, A.L., and Albert, R. (1999). Emergence of scaling in random networks. Science 286, 509–512. Abstract Article
Baugh, L.R., Hill, A.A., Claggett, J.M., Hill-Harfe, K., Wen, J.C., Slonim, D.K., Brown, E.L., and Hunter, C.P. (2005). The homeodomain protein PAL-1 specifies a lineage-specific regulatory network in the C. elegans embryo. Development 132, 1843–1854. Abstract Article
Baugh, L.R., Hill, A.A., Slonim, D.K., Brown, E.L., and Hunter, C.P. (2003). Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development 130, 889–900. Abstract Article
Beer, M.A., and Tavazoie, S. (2004). Predicting gene expression from sequence. Cell 117, 185–198. Abstract Article
Bertone, P., Stolc, V., Royce, T.E., Rozowsky, J.S., Urban, A.E., Zhu, X., Rinn, J.L., Tongprasit, W., Samanta, M., Weissman, S., et al. (2004). Global identification of human transcribed sequences with genome tiling arrays. Science 306, 2242–2246. Abstract Article
Boulton, S.J., Gartner, A., Reboul, J., Vaglio, P., Dyson, N., Hill, D.E., and Vidal, M. (2002). Combined functional genomic maps of the C. elegans DNA damage response. Science 295, 127–131. Abstract Article
Brasch, M.A., Hartley, J.L., and Vidal, M. (2004). ORFeome cloning and systems biology: standardized mass production of the parts from the parts-list. Genome Res. 14, 2001–2009. Abstract Article
Bustin, S.A. (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25, 169–193. Abstract Article
Carninci, P., Waki, K., Shiraki, T., Konno, H., Shibata, K., Itoh, M., Aizawa, K., Arakawa, T., Ishii, Y., Sasaki, D., et al. (2003). Targeting a complex transcriptome: the construction of the mouse full-length cDNA encyclopedia. Genome Res. 13, 1273–1289. Abstract Article
Castelli, V., Aury, J.M., Jaillon, O., Wincker, P., Clepet, C., Menard, M., Cruaud, C., Quetier, F., Scarpelli, C., Schachter, V., et al. (2004). Whole genome sequence comparisons and “full-length” cDNA sequences: a combined approach to evaluate and improve Arabidopsis genome annotation. Genome Res. 14, 406–413. Abstract Article
Castillo-Davis, C.I., Hartl, D.L., and Achaz, G. (2004). cis-Regulatory and protein evolution in orthologous and duplicate genes. Genome Res. 14, 1530–1536. Abstract Article
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W.W., and Prasher, D.C. (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. Abstract
Chen, N., Harris, T.W., Antoshechkin, I., Bastiani, C., Bieri, T., Blasiar, D., Bradnam, K., Canaran, P., Chan, J., Chen, C.K., et al. (2005). WormBase: a comprehensive data resource for Caenorhabditis biology and genomics. Nucleic Acids Res. 33, D383–D389. Abstract Article
Cinar, H., Keles, S., and Jin, Y. (2005). Expression Profiling of GABAergic Motor Neurons in Caenorhabditis elegans. Curr. Biol. 15, 340–346. Abstract Article
Colaiacovo, M.P., Stanfield, G.M., Reddy, K.C., Reinke, V., Kim, S.K., and Villeneuve, A.M. (2002). A targeted RNAi screen for genes involved in chromosome morphogenesis and nuclear organization in the Caenorhabditis elegans germline. Genetics 162, 113–128. Abstract
Colosimo, M.E., Brown, A., Mukhopadhyay, S., Gabel, C., Lanjuin, A.E., Samuel, A.D., and Sengupta, P. (2004). Identification of thermosensory and olfactory neuron-specific genes via expression profiling of single neuron types. Curr. Biol. 14, 2245–2251. Abstract Article
Das, M., Harvey, I., Chu, L.L., Sinha, M., and Pelletier, J. (2001). Full-length cDNAs: more than just reaching the ends. Physiol. Genomics 6, 57–80. Abstract
Davy, A., Bello, P., Thierry-Mieg, N., Vaglio, P., Hitti, J., Doucette-Stamm, L., Thierry-Mieg, D., Reboul, J., Boulton, S., Walhout, A.J., et al. (2001). A protein-protein interaction map of the Caenorhabditis elegans 26S proteasome. EMBO Rep. 2, 821–828. Abstract Article
Denver, D.R., Morris, K., Streelman, J.T., Kim, S.K., Lynch, M., and Thomas, W.K. (2005). The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat. Genet. 37, 544–548. Abstract Article
Deplancke, B., Dupuy, D., Vidal, M., and Walhout, A.J. (2004). A gateway-compatible yeast one-hybrid system. Genome Res. 14, 2093–2101. Abstract Article
Deplancke, B., Mukhopadhyay, A., Ao, W., Elewa, A.M., Grove, C.A., Martinez, N.J., Sequerra, R., Doucette-Stamm, L., Reece-Hoyes, J.S., Hope, I.A., Tissenbaum, H.A., Mango, S.E., and Walhout, A.J. (2006). A gene-centered C. elegans protein-DNA interaction network. Cell 125, 1032–1034. Abstract Article
Dinkova, T.D., Keiper, B.D., Korneeva, N.L., Aamodt, E.J., and Rhoads, R.E. (2005). Translation of a small subset of Caenorhabditis elegans mRNAs is dependent on a specific eukaryotic translation initiation factor 4E isoform. Mol. Cell. Biol. 25, 100–113. Abstract Article
Dupuy, D., Li, Q.R., Deplancke, B., Boxem, M., Hao, T., Lamesch, P., Sequerra, R., Bosak, S., Doucette-Stamm, L., Hope, I.A., et al. (2004). A first version of the Caenorhabditis elegans Promoterome. Genome Res. 14, 2169–2175. Abstract Article
Durbin, R., and Thierry-Mieg, J. (1994). The ACeDB genome database. In: Computational Methods in Genome Research, Suhai, S., ed., New York, NY: Plenum Press, pp 45–56.
Endoh, H., Vincent, S., Jacob, Y., Real, E., Walhout, A.J., and Vidal, M. (2002). Integrated version of reverse two-hybrid system for the postproteomic era. Meth. Enzymol. 350, 525–545. Abstract
Endoh, H., Walhout, A.J., and Vidal, M. (2000). A green fluorescent protein-based reverse two-hybrid system: application to the characterization of large numbers of potential protein-protein interactions. Meth. Enzymol. 328, 74–88. Abstract
Feng, Z., Cronin, C.J., Wittig, J.H., Jr., Sternberg, P.W., and Schafer, W.R. (2004). An imaging system for standardized quantitative analysis of C. elegans behavior. BMC Bioinformatics 5, 115. Abstract Article
Fernandez, A.G., Gunsalus, K.C., Huang, J., Chuang, L.S., Ying, N., Liang, H.L., Tang, C., Schetter, A.J., Zegar, C., Rual, J.F., et al. (2005). New genes with roles in the C. elegans embryo revealed using RNAi of ovary-enriched ORFeome clones. Genome Res. 15, 250–259. Abstract Article
Fields, S., and Song, O. (1989). A novel genetic system to detect protein-protein interactions. Nature 340, 245–246. Abstract Article
Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. Abstract Article
Fox, R.M., Von Stetina, S.E., Barlow, S.J., Shaffer, C., Olszewski, K.L., Moore, J.H., Dupuy, D., Vidal, M., and Miller, D.M., 3rd (2005). A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics 6, 42. Abstract Article
Fraser, A.G., Kamath, R.S., Zipperlen, P., Martinez-Campos, M., Sohrmann, M., and Ahringer, J. (2000). Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408, 325–330. Abstract Article
Ge, H., Liu, Z., Church, G.M., and Vidal, M. (2001). Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 29, 482–486. Abstract Article
Ge, H., Walhout, A.J., and Vidal, M. (2003). Integrating 'omic' information: a bridge between genomics and systems biology. Trends Genet. 19, 551–560. Abstract Article
Geng, W., Cosman, P., Baek, J.H., Berry, C.C., and Schafer, W.R. (2003). Quantitative classification and natural clustering of Caenorhabditis elegans behavioral phenotypes. Genetics 165, 1117–1126. Abstract
Geng, W., Cosman, P., Berry, C.C., Feng, Z., and Schafer, W.R. (2004). Automatic tracking, feature extraction and classification of C elegans phenotypes. IEEE Trans. Biomed. Eng. 51, 1811–1820. Abstract Article
Giaever, G., Chu, A.M., Ni, L., Connelly, C., Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., Andre, B., et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. Abstract Article
Goldberg, D.S., and Roth, F.P. (2003). Assessing experimentally derived interactions in a small world. Proc. Natl. Acad. Sci. U.S.A. 100, 4372–4376. Abstract Article
Gönczy, P., Echeverri, G., Oegema, K., Coulson, A., Jones, S.J., Copley, R.R., Duperon, J., Oegema, J., Brehm, M., Cassin, E., et al. (2000). Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature 408, 331–336. Abstract Article
Grigoriev, A. (2001). A relationship between gene expression and protein interactions on the proteome scale: analysis of the bacteriophage T7 and the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 29, 3513–3519. Abstract Article
GuhaThakurta, D., Palomar, L., Stormo, G.D., Tedesco, P., Johnson, T.E., Walker, D.W., Lithgow, G., Kim, S., and Link, C.D. (2002). Identification of a novel cis-regulatory element involved in the heat shock response in Caenorhabditis elegans using microarray gene expression and computational methods. Genome Res. 12, 701–712. Abstract Article
Gunsalus, K.C., Ge, H., Schetter, A.J., Goldberg, D.S., Han, J.D., Hao, T., Berriz, G.F., Bertin, N., Huang, J., Chuang, L.S., et al. (2005). Predictive models of molecular machines involved in Caenorhabditis elegans early embryogenesis. Nature 436, 861–865. Abstract Article
Gunsalus, K.C., and Piano, F. (2005). RNAi as a tool to study cell biology: building the genome-phenome bridge. Curr. Opin. Cell Biol. 17, 3–8. Abstract Article
Gunsalus, K.C., Yueh, W.C., MacMenamin, P., and Piano, F. (2004). RNAiDB and PhenoBlast: web tools for genome-wide phenotypic mapping projects. Nucleic Acids Res. 32, D406–D410. Abstract Article
Guo, S., and Kemphues, K.J. (1995). par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell 81, 611–620. Abstract Article
Han, J.D., Bertin, N., Hao, T., Goldberg, D.S., Berriz, G.F., Zhang, L.V., Dupuy, D., Walhout, A.J., Cusick, M.E., Roth, F.P., and Vidal, M. (2004). Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93. Abstract Article
Hanlon, S.E., and Lieb, J.D. (2004). Progress and challenges in profiling the dynamics of chromatin and transcription factor binding with DNA microarrays. Curr. Opin. Genet. Dev. 14, 697–705. Abstract Article
Hartwell, L.H., Hopfield, J.J., Leibler, S., and Murray, A.W. (1999). From molecular to modular cell biology. Nature 402, C47–C52. Abstract Article
Hill, A.A., Hunter, C.P., Tsung, B.T., Tucker-Kellogg, G., and Brown, E.L. (2000). Genomic analysis of gene expression in C. elegans. Science 290, 809–812. Abstract Article
Hillier, L.W., Coulson, A., Murray, J.I., Bao, Z., Sulston, J.E., and Waterston, R.H. (2005). Genomics in C. elegans: so many genes, such a little worm. Genome Res. 15, 1651–1660. Abstract Article
Holt, S.J., and Riddle, D.L. (2003). SAGE surveys C. elegans carbohydrate metabolism: evidence for an anaerobic shift in the long-lived dauer larva. Mech. Ageing Dev. 124, 779–800. Abstract Article
Hope, I.A., Albertson, D.G., Martinelli, S.D., Lynch, A.S., Sonnhammer, E., and Durbin, R. (1996). The C. elegans expression pattern database: a beginning. Trends Genet. 12, 370–371. Abstract Article
Hope, I.A., Stevens, J., Garner, A., Hayes, J., Cheo, D.L., Brasch, M.A., and Vidal, M. (2004). Feasibility of genome-scale construction of promoter::reporter gene fusions for expression in Caenorhabditis elegans using a multisite gateway recombination system. Genome Res. 14, 2070–2075. Abstract Article
Jansen, R., Greenbaum, D., and Gerstein, M. (2002). Relating whole-genome expression data with protein-protein interactions. Genome Res. 12, 37–46. Abstract Article
Jeong, H., Mason, S.P., Barabasi, A.L., and Oltvai, Z.N. (2001). Lethality and centrality in protein networks. Nature 411, 41–42. Abstract Article
Jiang, M., Ryu, J., Kiraly, M., Duke, K., Reinke, V., and Kim, S.K. (2001). Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 98, 218–223. Abstract Article
Jones, S.J., Riddle, D.L., Pouzyrev, A.T., Velculescu, V.E., Hillier, L., Eddy, S.R., Stricklin, S.L., Baillie, D.L., Waterston, R., and Marra, M.A. (2001). Changes in gene expression associated with developmental arrest and longevity in Caenorhabditis elegans. Genome Res. 11, 1346–1352. Abstract Article
Kamath, R.S., Fraser, A.G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. Abstract Article
Kemmeren, P., van Berkum, N.L., Vilo, J., Bijma, T., Donders, R., Brazma, A., and Holstege, F.C. (2002). Protein interaction verification and functional annotation by integrated analysis of genome-scale data. Mol. Cell 9, 1133–1143. Abstract Article
Kent, W.J., and Zahler, A.M. (2000). The intronerator: exploring introns and alternative splicing in Caenorhabditis elegans. Nucleic Acids Res. 28, 91–93. Abstract Article
Kim, S.K., Lund, J., Kiraly, M., Duke, K., Jiang, M., Stuart, J.M., Eizinger, A., Wylie, B.N., and Davidson, G.S. (2001). A gene expression map for Caenorhabditis elegans. Science 293, 2087–2092. Abstract Article
Kniazeva, M., Crawford, Q.T., Seiber, M., Wang, C.Y., and Han, M. (2004). Monomethyl branched-chain fatty acids play an essential role in Caenorhabditis elegans development. PLoS Biol. 2, E257. Abstract Article
Korf, I., Flicek, P., Duan, D., and Brent, M.R. (2001). Integrating genomic homology into gene structure prediction. Bioinformatics 17, S140–S148. Abstract
Kunitomo, H., Uesugi, H., Kohara, Y., and Iino, Y. (2005). Identification of ciliated sensory neuron-expressed genes in Caenorhabditis elegans using targeted pull-down of poly(A) tails. Genome Biol. 6, R17. Abstract Article
Kwon, J.Y., Hong, M., Choi, M.S., Kang, S., Duke, K., Kim, S., Lee, S., and Lee, J. (2004). Ethanol-response genes and their regulation analyzed by a microarray and comparative genomic approach in the nematode Caenorhabditis elegans. Genomics 83, 600–614. Abstract Article
Lamesch, P., Milstein, S., Hao, T., Rosenberg, J., Li, N., Sequerra, R., Bosak, S., Doucette-Stamm, L., Vandenhaute, J., Hill, D.E., and Vidal, M. (2004). C. elegans ORFeome version 3.1: increasing the coverage of ORFeome resources with improved gene predictions. Genome Res. 14, 2064–2069. Abstract Article
Lamitina, S.T., and Strange, K. (2005). Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am. J. Physiol., Cell Physiol. 288, C467–C474. Abstract Article
Li, S., Armstrong, C.M., Bertin, N., Ge, H., Milstein, S., Boxem, M., Vidalain, P.O., Han, J.D., Chesneau, A., Hao, T., et al. (2004). A map of the interactome network of the metazoan C. elegans. Science 303, 540–543. Abstract Article
Link, C.D., Taft, A., Kapulkin, V., Duke, K., Kim, S., Fei, Q., Wood, D.E., and Sahagan, B.G. (2003). Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer's disease model. Neurobiol. Aging 24, 397–413. Abstract Article
Lund, J., Tedesco, P., Duke, K., Wang, J., Kim, S.K., and Johnson, T.E. (2002). Transcriptional profile of aging in C. elegans. Curr. Biol. 12, 1566–1573. Abstract Article
Maeda, I., Kohara, Y., Yamamoto, M., and Sugimoto, A. (2001). Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol. 11, 171–176. Abstract Article
McCarroll, S.A., Li, H., and Bargmann, C.I. (2005). Identification of transcriptional regulatory elements in chemosensory receptor genes by probabilistic segmentation. Curr. Biol. 15, 347–352. Abstract Article
McCarroll, S.A., Murphy, C.T., Zou, S., Pletcher, S.D., Chin, C.S., Jan, Y.N., Kenyon, C., Bargmann, C.I., and Li, H. (2004). Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat. Genet. 36, 197–204. Abstract Article
McElwee, J.J., Schuster, E., Blanc, E., Thomas, J.H., and Gems, D. (2004). Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 279, 44533–44543. Abstract Article
McKay, S.J., Johnsen, R., Khattra, J., Asano, J., Baillie, D.L., Chan, S., Dube, N., Fang, L., Goszczynski, B., Ha, E., et al. (2003). Gene expression profiling of cells, tissues, and developmental stages of the nematode C. elegans. Cold Spring Harb. Symp. Quant. Biol. 68, 159–169. Abstract Article
Mohler, W., Millard, A.C., and Campagnola, P.J. (2003). Second harmonic generation imaging of endogenous structural proteins. Methods 29, 97–109. Abstract Article
Mohler, W.A., and White, J.G. (1998). Stereo-4-D reconstruction and animation from living fluorescent specimens. Biotechniques 24, 1006–1010, 1012. Abstract
Nelson, G.A., Jones, T.A., Chesnut, A., and Smith, A.L. (2002). Radiation-induced gene expression in the nematode Caenorhabditis elegans. J. Radiat. Res. 43, S199–S203. Abstract Article
Ning, F., LeCun, Y., Piano, F., Bottou, L., and Barbano, P.E. (2005). Toward Automatic Phenotyping of Developing Embryos from Videos. IEEE Trans. Image Process. (in press).
Oh, S.W., Mukhopadhyay, A., Dixit, B.L., Raha, T., Green, M.R., and Tissenbaum, H.A. (2006). Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat. Genet. 38, 251–257. Abstract Article
Owen, A.B., Stuart, J., Mach, K., Villeneuve, A.M., and Kim, S. (2003). A gene recommender algorithm to identify coexpressed genes in C. elegans. Genome Res. 13, 1828–1837. Abstract Article
Piano, F., Schetter, A.J., Mangone, M., Stein, L., and Kemphues, K.J. (2000). RNAi analysis of genes expressed in the ovary of Caenorhabditis elegans. Curr. Biol. 10, 1619–1622. Abstract Article
Piano, F., Schetter, A.J., Morton, D.G., Gunsalus, K.C., Reinke, V., Kim, S.K., and Kemphues, K.J. (2002). Gene Clustering Based on RNAi Phenotypes of Ovary-Enriched Genes in C. elegans. Curr. Biol. 12, 1959–1964. Abstract Article
Pleasance, E.D., Marra, M.A., and Jones, S.J. (2003). Assessment of SAGE in transcript identification. Genome Res. 13, 1203–1215. Abstract Article
Qiu, S., Adema, C.M., and Lane, T. (2005). A computational study of off-target effects of RNA interference. Nucleic Acids Res. 33, 1834–1847. Abstract Article
Reboul, J., Vaglio, P., Rual, J.F., Lamesch, P., Martinez, M., Armstrong, C.M., Li, S., Jacotot, L., Bertin, N., Janky, R., et al. (2003). C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat. Genet. 34, 35–41. Abstract Article
Reboul, J., Vaglio, P., Tzellas, N., Thierry-Mieg, N., Moore, T., Jackson, C., Shin-i, T., Kohara, Y., Thierry-Mieg, D., Thierry-Mieg, J., et al. (2001). Open-reading-frame sequence tags (OSTs) support the existence of at least 17,300 genes in C. elegans. Nat. Genet. 27, 332–336. Abstract Article
Reinke, V., Gil, I.S., Ward, S., and Kazmer, K. (2004). Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 131, 311–323. Abstract Article
Reinke, V., Smith, H.E., Nance, J., Wang, J., Van Doren, C., Begley, R., Jones, S.J., Davis, E.B., Scherer, S., Ward, S., and Kim, S.K. (2000). A global profile of germline gene expression in C. elegans. Mol. Cell 6, 605–616. Abstract Article
Robertson, S.M., Shetty, P., and Lin, R. (2004). Identification of lineage-specific zygotic transcripts in early Caenorhabditis elegans embryos. Dev. Biol. 276, 493–507. Abstract Article
Roy, P.J., Stuart, J.M., Lund, J., and Kim, S.K. (2002). Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418, 975–979. Abstract Article
Rual, J.F., Ceron, J., Koreth, J., Hao, T., Nicot, A.S., Hirozane-Kishikawa, T., Vandenhaute, J., Orkin, S.H., Hill, D.E., van den Heuvel, S., and Vidal, M. (2004). Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 14, 2162–2168. Abstract Article
Said, M.R., Begley, T.J., Oppenheim, A.V., Lauffenburger, D.A., and Samson, L.D. (2004). Global network analysis of phenotypic effects: protein networks and toxicity modulation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 101, 18006–18011. Abstract Article
SenGupta, D.J., Zhang, B., Kraemer, B., Pochart, P., Fields, S., and Wickens, M. (1996). A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 93, 8496–8501. Abstract Article
Simmer, F., Moorman, C., van der Linden, A.M., Kuijk, E., van den Berghe, P.V., Kamath, R.S., Fraser, A.G., Ahringer, J., and Plasterk, R.H. (2003). Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 1, E12. Abstract Article
Sönnichsen, B., Koski, L.B., Walsh, A., Marschall, P., Neumann, B., Brehm, M., Alleaume, A.M., Artelt, J., Bettencourt, P., Cassin, E., et al. (2005). Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature 434, 462–469. Abstract Article
Stein, L., Sternberg, P., Durbin, R., Thierry-Mieg, J., and Spieth, J. (2001). WormBase: network access to the genome and biology of Caenorhabditis elegans. Nucleic Acids Res. 29, 82–86. Abstract Article
Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al. (2003). The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1, E45. Abstract Article
Stolc, V., Gauhar, Z., Mason, C., Halasz, G., van Batenburg, M.F., Rifkin, S.A., Hua, S., Herreman, T., Tongprasit, W., Barbano, P.E., et al. (2004). A gene expression map for the euchromatic genome of Drosophila melanogaster. Science 306, 655–660. Abstract Article
Stuart, J.M., Segal, E., Koller, D., and Kim, S.K. (2003). A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255. Abstract Article
Sulston, J.E., and Horvitz, H.R. (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156. Abstract Article
Sulston, J.E., Schierenberg, E., White, J.G., and Thomson, J.N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119. Abstract Article
Tewari, M., Hu, P.J., Ahn, J.S., Ayivi-Guedehoussou, N., Vidalain, P.O., Li, S., Milstein, S., Armstrong, C.M., Boxem, M., Butler, M.D., et al. (2004). Systematic interactome mapping and genetic perturbation analysis of a C. elegans TGF-beta signaling network. Mol. Cell 13, 469–482. Abstract Article
The C. elegans Sequencing Consortium (1998). Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018. Abstract Article
Thierry-Mieg, J., Thierry-Mieg, D., Potdevin, M., and Sienkiewicz, M. (2005). Identification and functional annotation of cDNA-supported genes in higher organisms using AceView. (http://www.wormgenes.org)
Vidal, M., Brachmann, R.K., Fattaey, A., Harlow, E., and Boeke, J.D. (1996). Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 93, 10315–10320. Abstract Article
Vidal, M., Braun, P., Chen, E., Boeke, J.D., and Harlow, E. (1996). Genetic characterization of a mammalian protein-protein interaction domain by using a yeast reverse two-hybrid system. Proc. Natl. Acad. Sci. U.S.A. 93, 10321–10326. Abstract Article
Vidal, M., and Legrain, P. (1999). Yeast forward and reverse 'n'-hybrid systems. Nucleic Acids Res. 27, 919–929. Abstract Article
Vidalain, P.O., Boxem, M., Ge, H., Li, S., and Vidal, M. (2004). Increasing specificity in high-throughput yeast two-hybrid experiments. Methods 32, 363–370. Abstract Article
Walhout, A.J., Boulton, S.J., and Vidal, M. (2000). Yeast two-hybrid systems and protein interaction mapping projects for yeast and worm. Yeast 17, 88–94. Abstract
Walhout, A.J., Reboul, J., Shtanko, O., Bertin, N., Vaglio, P., Ge, H., Lee, H., Doucette-Stamm, L., Gunsalus, K.C., Schetter, A.J., et al. (2002). Integrating interactome, phenome, and transcriptome mapping data for the C. elegans germline. Curr. Biol. 12, 1952–1958. Abstract Article
Walhout, A.J., Sordella, R., Lu, X., Hartley, J.L., Temple, G.F., Brasch, M.A., Thierry-Mieg, N., and Vidal, M. (2000). Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 287, 116–122. Abstract Article
Walhout, A.J., Temple, G.F., Brasch, M.A., Hartley, J.L., Lorson, M.A., van den Heuvel, S., and Vidal, M. (2000). GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Meth. Enzymol. 328, 575–592. Abstract
Walhout, A.J., and Vidal, M. (2001). High-throughput yeast two-hybrid assays for large-scale protein interaction mapping. Methods 24, 297–306. Abstract Article
Walhout, M., Endoh, H., Thierry-Mieg, N., Wong, W., and Vidal, M. (1998). A model of elegance. Am. J. Hum. Genet. 63, 955–961. Abstract Article
Wang, J., and Kim, S.K. (2003). Global analysis of dauer gene expression in Caenorhabditis elegans. Development 130, 1621–1634. Abstract Article
Washington University Genome Sequencing Center (2005). C. elegans long-oligomer spotted arrays. (http://www.genome.wustl.edu/)
Waterston, R., Martin, C., Craxton, M., Huynh, C., Coulson, A., Hillier, L., Durbin, R., Green, P., Shownkeen, R., Halloran, N., and et al. (1992). A survey of expressed genes in Caenorhabditis elegans. Nat. Genet. 1, 114–123. Abstract Article
Wei, C., Lamesch, P., Arumugam, M., Rosenberg, J., Hu, P., Vidal, M., and Brent, M.R. (2005). Closing in on the C. elegans ORFeome by cloning TWINSCAN predictions. Genome Res. 15, 577–582. Abstract Article
White, J.G., Horvitz, H.R., and Sulston, J.E. (1982). Neurone differentiation in cell lineage mutants of Caenorhabditis elegans. Nature 297, 584–587. Abstract Article
Winzeler, E.A., Shoemaker, D.D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J.D., Bussey, H., et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. Abstract Article
Yamada, K., Lim, J., Dale, J.M., Chen, H., Shinn, P., Palm, C.J., Southwick, A.M., Wu, H.C., Kim, C., Nguyen, M., et al. (2003). Empirical analysis of transcriptional activity in the Arabidopsis genome. Science 302, 842–846. Abstract Article
Yasuda, T., Bannai, H., Onami, S., Miyano, S., and Kitano, H. (1999). Towards Automatic Construction of Cell-Lineage of C. elegans from Nomarski DIC Microscope Images. Genome Inform. Ser. Workshop Genome Inform. 10, 144–154. Abstract
Zhang, S., Ma, C., and Chalfie, M. (2004). Combinatorial marking of cells and organelles with reconstituted fluorescent proteins. Cell 119, 137–144. Abstract Article
Zhang, Y., Ma, C., Delohery, T., Nasipak, B., Foat, B.C., Bounoutas, A., Bussemaker, H.J., Kim, S.K., and Chalfie, M. (2002). Identification of genes expressed in C. elegans touch receptor neurons. Nature 418, 331–335. Abstract Article
*Edited by Jonathan Hodgkin. Last revised May 21, 2006. Published August 21, 2006. This chapter should be cited as: Piano, F., et al. C. elegans network biology: a beginning (August 21, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.118.1, http://www.wormbook.org.
Copyright: © 2006 Fabio Piano, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: fp1@nyu.edu, kcg1@nyu.edu, david_hill@dfci.harvard.edu, or marc_vidal@ccsb.dfci.harvard.edu.
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.