Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
As in all living organisms, survival in C. elegans requires adequate management of energy supplies. Genetic screens have revealed that C. elegans fat regulation involves a complex network of genes with known or likely functions in food sensation, neuroendocrine signaling, uptake, transport, storage and utilization of fats. Core fat and sugar metabolic pathways are conserved in C. elegans. Flux through these pathways is modulated by cellular energy sensors that operate via transcriptional and translational regulatory mechanisms. In turn, neuroendocrine mechanisms couple sensory and metabolic pathways while neuromodulatory pathways influence both metabolic and food seeking/consumption pathways. The shared ancestry of C. elegans and mammalian fat regulatory pathways extends to developmental programs that underlie fat storage capacity, despite lack of dedicated adipocytes, and genes whose human homologs are implicated in obesity. This suggests that many of the newly identified C. elegans fat regulatory pathways play similar roles in mammals. C. elegans is ideally suited for the integrated study of mechanisms that operate in multiple tissues and elicit feedback responses that affect processes as diverse as metabolism and behavior.
Obesity is a significant risk factor for major diseases including Type II diabetes, coronary heart disease, hypertension and certain forms of cancer (Barsh et al., 2000; Kopelman, 2000; Luchsinger, 2006). Obesity arises when energy intake, principally stored as triglycerides, exceeds energy expenditure (Flier, 2004; Spiegelman and Flier, 2001). Obesity is a complex trait influenced by diet, developmental stage, age, physical activity and genes (Brockmann and Bevova, 2002; Friedman, 2003).
Genetic predisposition is a key contributing factor in obesity as demonstrated by familial aggregation, twin and adoption studies (Allison et al., 1996; Friedman, 2003; Stunkard et al., 1990). Estimates for the genetic basis of phenotypic variations in obesity range from approximately 40 to 70%. This matches or exceeds the accepted genetic contribution to height (Friedman, 2003). The idea that genetic loci alter body fat content has been substantiated by identification of mutations that cause low- or high-fat phenotypes in rodents and humans (Brockmann and Bevova, 2002; Delrue and Michaud, 2004).
There is convincing experimental evidence showing that the balance between energy intake (food consumption) and energy expenditure (basal metabolic rate, i.e. biochemical processes required to maintain cellular viability, physical activity and adaptive thermogenesis) is tightly regulated. A homeostatic network maintains energy stores through a complex interplay between the feeding regulatory centers in the central nervous system (CNS), particularly in the hypothalamus and the regulated storage and mobilization of fat stores (see Figure 1; Cone, 2005; Friedman, 2000, Flier, 2004; Sainsbury et al., 2002; Spiegelman and Flier, 2001). Thus, genes that encode the molecular components of this system may underlie obesity and related disorders.
|
Figure 1. Homeostatic regulation of energy balance in mammals. Signals from sites of fat storage communicate the energetic state of the body to the nervous system, which also receives environmental and sensory inputs. The nervous system integrates these signals and responds to alter behavior, physiology and energy uptake, storage and utilization.
In mammals, white adipose tissue functions as the main depot for fuel storage. In the past decade, identification of myriad lipid and protein signals secreted from this tissue has led to its recognition as a major endocrine organ (Rondinone, 2006; Trayhurn and Bing, 2006). This was principally based on the discovery of leptin, a cytokine-like hormone secreted from white adipose tissue in proportion to fat mass (Friedman and Halaas, 1998). Activation of hypothalamic leptin receptors suppresses food intake and promotes energy expenditure pathways (Friedman, 2002; Porte et al., 2002).
Insulin is another key afferent signal to the CNS that controls energy balance. Insulin is secreted from the endocrine pancreas in proportion to fat mass and exerts potent effects on peripheral nutrient storage. Similar to leptin, insulin causes long-term inhibitory effects on energy intake. There is cross talk between insulin and leptin signaling in a common set of hypothalamic neurons. Moreover, a series of neuropeptides (e.g., the melanocortin system, neuropeptide Y) and neurotransmitters (e.g., serotonin, dopamine and noradrenaline) function in the hypothalamus to coordinate behavioral, physiological and metabolic responses. Together, these responses maintain energy balance via both intake and expenditure pathways (Cone, 2005; Porte et al., 2002; Sainsbury et al., 2002).
In addition to these long-term adiposity signals, short-term meal-related signals are transmitted to the CNS through afferent nerves or gut-secreted peptides (e.g., cholecystokinin, ghrelin; Badman and Flier, 2005). Finally, neurons in the CNS also directly sense carbohydrate and fats (Demuro and Obici, 2006; Lam et al., 2005).
Because energy balance involves this complex interplay between multiple tissues and signaling pathways, an integrated view of feeding behavior, neuroendocrine signaling, nutrient uptake, transport, storage and utilization is required for understanding fat regulation. Moreover, developmental programs that underlie fat storage capacity are fundamental to understanding fat regulation. A wealth of genetic and behavioral tools makes C. elegans an excellent system for unraveling these complex pathways. Thus, in the past few years, the study of fat in C. elegans has emerged as an exciting field that is yielding new insights in the regulation of energy balance at the level of the whole organism.
Several groups have biochemically determined the composition of C. elegans fat content. This is accomplished by extraction of total lipids from whole animals, fractionation to phospholipids and neutral lipid moieties, and further analysis by column and thin-layer chromatography as well as gas-chromatography/mass spectrometry (GC/MS; Kniazeva et al., 2003; Satouchi et al., 1993; Watts and Browse, 2002). Triacylglyceride fat stores make up approximately 40–55% of total lipids depending on diet and growth stage (Ashrafi, 2006). Phospholipids pools are composed of approximately 55% ethanolamine glycerophospholipid, 32% choline glycerophospholipid, 8% sphingomyelin. Cardiolipin, inositol glycerophospholipids and lyso-cholineglycero- phospholipids account for the remaining 5%. Relative abundance of these phospholipid constituents changes with growth temperature (Satouchi et al., 1993; Tanaka et al., 1996).
Similar to mammals, C. elegans contains a wide range of saturated, monounsaturated and polyunsaturated fatty acids (PUFAs) including arachidonic (20:4n-6) and eicosapentaenoic acid (20:5n-3) as well as monomethyl branched chain fatty acids (mmBCFAs; Kniazeva et al., 2004; Satouchi et al., 1993; Watts and Browse, 2002). C. elegans combines the full range of desaturase and PUFA elongase activities that are found separately in plants and animals, abrogating the requirement for dietary supply of linoleic acid (18:2n6) and linolenic acid (18:3n3), which are essential fatty acids in mammalian diets (Brock et al., 2006; Wallis et al., 2002; Watts and Browse, 2002). Inactivation of C. elegans desaturase and elongase family members, encoded by fat and elo genes, respectively, causes imbalances in fatty acid composition and is associated with metabolic, physiological and behavioral phenotypes. These include altered total fat levels, growth retardation, slowed movement, reduction in body size, germ cell maintenance and reproductive defects, aberrations in rhythmic behavior, defects in sensory signaling, defects in neurotransmission and reduced adult lifespan (Brock et al., 2006; Kahn-Kirby et al., 2004; Kniazeva et al., 2004; Kniazeva et al., 2003; Lesa et al., 2003; Van Gilst et al., 2005; Watts and Browse, 2006; Watts et al., 2003). Some of the reported phenotypes may be indirect consequences of global alterations in fat levels and membrane composition.
Finally, C. elegans are cholesterol auxotrophs requiring dietary supply of this sterol. Given the small quantities of cholesterol needed for viability, it has been postulated that cholesterol functions as a precursor for sterol-derived hormones rather than playing a structural role in membrane composition and fluidity (Kurzchalia and Ward, 2003).
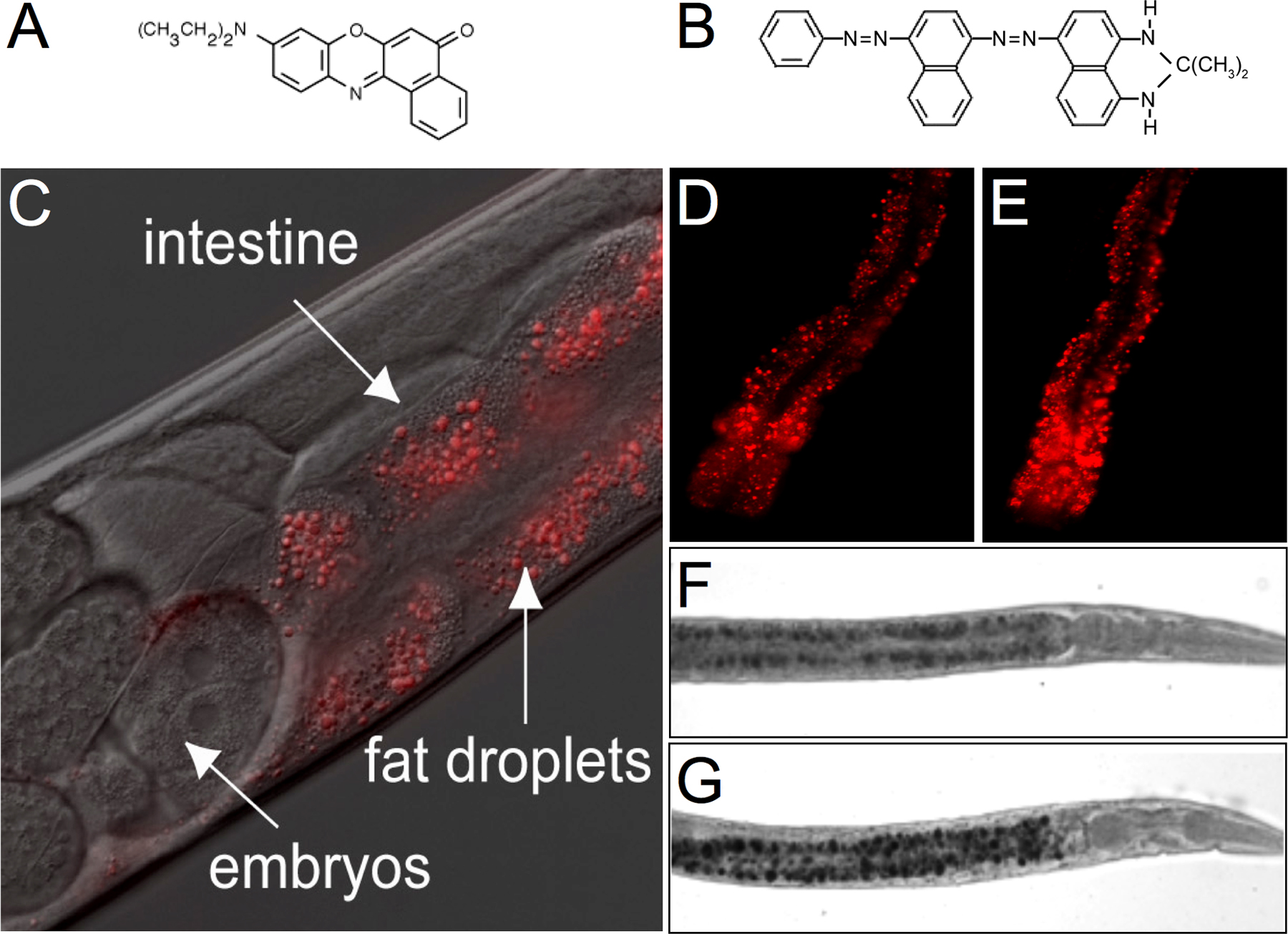
Whereas mammals have dedicated adipocytes, C. elegans store fat in droplets in their intestinal cells and in their hypodermal cells (see Figure 2). Because C. elegans have transparent bodies, these fat stores can be directly visualized in intact animals. A classic method is to stain fixed animals with the fat-soluble dye Sudan Black B (see Figure 2F and G; Kimura et al., 1997), which produces a blue-black stain visible under a standard dissection microscope.
|
Figure 2. Visualization of intestinal lipid droplets in transparent bodies of C. elegans. (A-B) Chemical structures of Nile Red (A) and Sudan Black (B). (C-E) Nile Red staining in wild-type N2 animals (C, D) and tub-1(nr2004) mutants (E). In panels D and E, the head of the animal is toward the bottom of the panel. (F-G) Sudan Black staining in wild-type N2 animals (F) and TGF-β receptor daf-1 (m40) mutants. The head of the animal is to the right.
Intestinal lipid droplets can also be visualized by supplementing the normal laboratory diet of C. elegans with Nile Red or BODIPY-labeled fatty acids (4,4-difluoro-5-methyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoic acid and 4,4-difluoro-5-octoyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoic acid; Figure 2C–E; Ashrafi et al., 2003). These dyes have greatly facilitated the genetic analysis of fat regulation as they allow for easy visualization of lipids in living nematodes. These compounds are brightly fluorescent and, have been used to visualize lipid droplets in cultured mammalian cells. These vital dyes do not produce any adverse effects on C. elegans growth rate, brood-size, pharyngeal pumping, dauer (a larval hibernation state) formation and recovery or lifespan. One limitation is that Nile Red and BODIPY-labeled fatty acid staining methods do not distinguish between animals with reduced fat levels and animals that fail to uptake these compounds. In such cases, Sudan Black B staining is the preferred method.
Fat visualization as well as direct examination of fat composition by GC/MS have formed the basis of genetic screens for identifying C. elegans fat regulatory pathways (Ashrafi et al., 2003; McKay et al., 2003; Watts and Browse, 2002). Unlike the GC/MS method, Sudan Black B., Nile Red and BODIPY-labeled fatty acids do not distinguish between different lipid compositions as these dyes have a general preference for hydrophobic moieties.
Targeted gene deletions, mutagenesis screens and a genome-scale RNA interference (RNAi) screen have identified approximately 300 gene inactivations that cause fat reduction and approximately 100 gene inactivations that cause fat accumulation without significant effects on growth and viability (Ashrafi et al., 2003; Jia et al., 2004; Kniazeva et al., 2004; Kniazeva et al., 2003; Ludewig et al., 2004; Mak et al., 2006; McKay et al., 2003; Mukhopadhyay et al., 2005; Taubert et al., 2006; Van Gilst et al., 2005; Vellai et al., 2003; Watts and Browse, 2002; Yang et al., 2006). Another approximately 250 gene inactivations cause dramatic fat reductions concomitant with defects ranging from sterility to growth arrest and lethality. Because of these pleiotropies, it is difficult to assign specific fat regulatory functions to such genes although they include some well-known components of metabolism.
The analysis of the genes identified through these screens is still in its infancy; however, recent reports demonstrate that they regulate fat content through diverse physiological processes. The shared ancestry of the mammalian and C. elegans fat regulatory pathways is highlighted in the sections below.
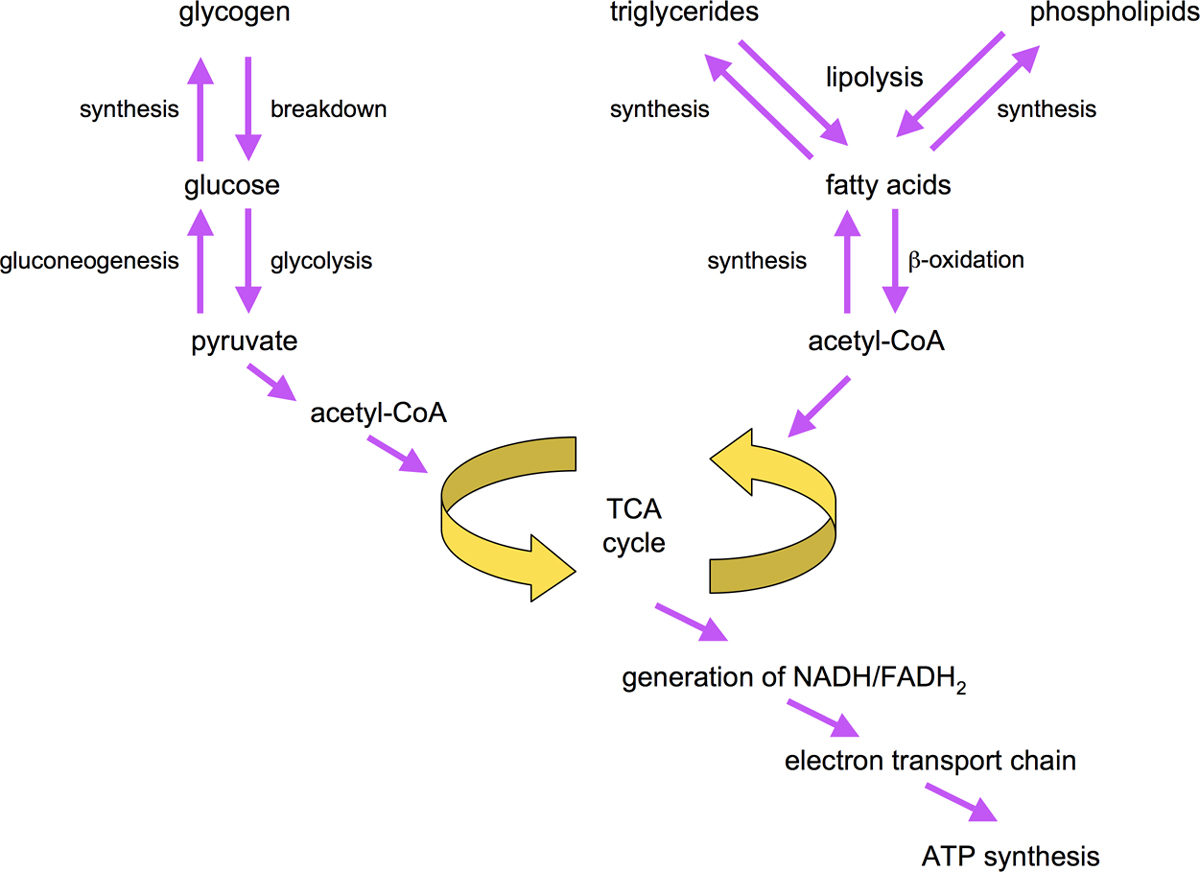
Intricate metabolic networks tightly coordinate the flow of sugars and fats through synthesis, storage, and breakdown pathways. These pathways are summarized in Figure 3.
In general, cells break down carbohydrates, amino acids and fats to generate ATP, the universal energy resource of cells (Salway, 2004). Carbohydrates are broken down via glycolytic enzymes to pyruvate and further to acetylCoA which powers generation of NADH and FADH2 through the tricarboxylic acid cycle (TCA) cycle. In turn, NADH and FADH2 are used to generate ATP via oxidative phosphorylation and ATP synthesis. Mobilization of stored triacylglycerides is initiated by lipolytic enzymes such as hormone-sensitive lipase. Liberated fatty acids are then activated to their respective acyl-CoA derivates by acyl-CoA synthases/ligases. Breakdown of fatty acyl-CoAs to acetyl-CoA occurs in peroxisomes or mitochondria via β-oxidation enzymes (Salway, 2004; Figure 3).
Acetyl-CoA is the key substrate for synthesis of fatty acids. Acetyl-CoA is carboxylated by acetyl-CoA carboxylase (ACC) to form malonyl-CoA, which is then elongated by fatty acid synthase (FAS) in a step-wise fashion to generate fatty acids of different lengths, mainly C16:0. Products of FAS are then acted upon by fatty acid desaturases to generate unsaturated fatty acids (see Figure 3 and Figure 5). Storage of fatty acids involves the step-wise conversion of fatty acyl-CoAs derived from exogenous or endogenous sources to phosphotidic acid, diacylglycerol and ultimately triacylglycerols (see Figure 3; Salway, 2004).
Glucose is synthesized by gluconeogenic enzymes. One substrate for gluconeogenesis is glycerol, which can be derived from breakdown of triacylglycerides. Depending on tissue, excess carbohydrates may be stored as glycogen. Alternatively, they are broken down to acetyl-CoA by glycolysis, and then converted to and stored as fats. Moreover, the glyoxylate pathway allows the interconversion of carbohydrates and fats through components of the TCA cycle.
The C. elegans genome encodes proteins with sequence homology to conserved components of carbohydrate and lipid synthesis and breakdown. Annotations of metabolic pathways are found at KEGG and Reactome databases. A partial list is presented in Table 1.
Table 1. Partial listing of C. elegans metabolic pathways deduced from the genome sequence.
| Carbohydrate metabolism | Lipid metabolism |
| Glycolysis/Gluconeogenesis | Lipolysis (hormone sensitive lipase) |
| Glycogen synthesis/Glyogen breakdown | Carnitine shuttle (fatty acid uptake) |
| Trehalose synthesis/Trehalose breakdown | Mitochondrial β-oxidation |
| Galactose metabolism | Peroxisomal β-oxidation |
| Fructose and mannose metabolism | Glycerol catabolism |
| Glyoxylate pathway | Fatty acid synthesis |
| Citric acid (TCA) cycle | Fatty acid elongation and desaturation |
| Triacylglyceride synthesis | |
| Energy metabolism | Phospholipids biosynthesis |
| Oxidative phosphorylation | Synthesis and utilization of ketone bodies |
| ATP synthesis | Sphingolipid and ceramide synthesis |
C. elegans metabolic pathways have been examined most extensively in the context of insulin signaling. This is because down-regulation of insulin signaling confers an extended adult lifespan as well as promoting dauer formation, the larval hibernation stage (see aging and dauer chapters in Post-embryonic development section of WormBook). Increased fat accumulation and altered metabolism are hallmarks of the long-lived, stress resistant dauers. Similarly, loss of function of daf-2, the C. elegans insulin-receptor, causes fat accumulation in adults (see Figure 4). Measurements of metabolic rate, as assessed by CO2 release, biochemical activity assessment of several key enzymes, microarray and serial analysis of gene expression, have all indicated global shifts in metabolic pathways associated with dauer larvae and daf-2 mutant adults (Braeckman et al., 2002; Burnell et al., 2005; Gems, 1999; Halaschek-Wiener et al., 2005; Holt and Riddle, 2003; Jones et al., 2001; Larsen et al., 1995; Lee et al., 2003; Lund et al., 2002; McElwee et al., 2004; McElwee et al., 2006; Murphy et al., 2003; Van Voorhies and Ward, 1999; Wadsworth and Riddle, 1989; Wang and Kim, 2003). In general, these shifts are reminiscent of metabolic adjustments observed in nutrient deprived or fasting mammals. These adjustments favor energy conservation, fat storage, and utilization of stored reservoirs. One complexity in interpreting these studies is that they analyze mRNAs and proteins extracted from whole animals. Since different tissues play different roles in energy balance, it is likely that identical metabolic pathways are modulated differentially in separate tissues.
|
Figure 4. Regulation of growth and metabolism by insulin signaling in C. elegans. Nutrients activate signaling by the DAF-2 insulin receptor to inhibit the FOXO-transcription factor DAF-16. This promotes growth and reproduction. Nutrient limitation down regulates signaling through the insulin receptor allowing activation of DAF-16. In an early larval stage, DAF-16 activity promotes dauer formation. In adults, DAF-16 reduces reproductive rate, enhances lifespan and causes fat accumulation. The C. elegans genome encodes numerous insulin-like molecules and those that signal nutritional availability are not known. Activated component of insulin signaling in different contexts of nutrient availability is shown in green.
Genetic alterations of metabolic enzymes profoundly impact fat levels in C. elegans. For example, RNAi inactivation of key glycolic and gluconeogenic genes such as GAPDH, an insulin-regulated glycolytic enzyme, and PEPCK, a regulated enzyme of gluconeogenesis and glyceroneogenesis, cause fat reduction (Ashrafi et al., 2003). Similarly, inactivations of fatty acid synthesis (e.g., acetylCoA carboxylate and fatty acid synthase), phospholipid synthesis (e.g., serine palmitoyltransferase, choline/ethanoloaminephosphotransferase) and triglyceride synthesis (e.g., glycerol-3-phosphate acyltransferase) pathways cause reductions in lipid accumulation and, in some cases, are associated with growth arrest (see Figure 3 and Figure 5; Ashrafi et al., 2003). Inactivation of fatty acid oxidation genes causes either decreased or increased fat levels (Ashrafi et al., 2003; Mak et al., 2006; Van Gilst et al., 2005). The basis for this paradoxical result is not yet clear but likely reflects compensatory and homeostatic mechanisms. Not surprisingly, inactivation of oxidative phosphorylation and ATP synthesis components is generally associated with profound reductions in fat levels concomitant with growth defects (K. Ashrafi, unpublished observations).
Inhibition of fat-5, fat-6, and fat-7 genes encoding delta-9 fatty acid desaturation enzymes is associated with reduced fat levels. Interestingly, RNAi inactivation of fat-7 causes fat reduction and shortened lifespan, phenotypes not seen in a fat-7 deletion mutation (Brock et al., 2006; Van Gilst et al., 2005). This discrepancy may be explained by the observation that loss of function mutations in fat-6 or fat-7 cause compensatory transcriptional responses in the remaining delta-9 desaturase genes. Accordingly, triple fat-5; fat-6; fat-7 are embryonic lethal (Brock et al., 2006).
Mammalian delta-9 stearoyl-CoA desaturase-1 (SCD-1) has emerged as a therapeutic target for obesity and metabolic disorders. SCD-1 is a target of leptin signaling. Significantly, SCD-1 knock-out mice display dramatic reductions in adiposity on otherwise wild-type or leptin deficient (ob/ob) backgrounds (Cohen and Friedman, 2004; Cohen et al., 2003). SCD-1 deficiency promotes β-oxidation pathways and decreases lipogenesis in liver and skeletal muscle. One proposed mechanism is that SCD-1 inhibition results in accumulation of saturated fatty acylCoAs which cause feedback inhibition of acyl-CoA carboxylase (ACC), the rate-limiting enzyme of fatty acid synthesis. ACC inhibition results in reduced accumulation of its product, malonylCoA, which in turn, relieves inhibition of carnitine-palmityol-transferase (CPT) shuttle. This allows for transport of fats into mitochondria for breakdown via β-oxidation (see Figure 5; Cohen and Friedman, 2004; Cohen et al., 2003). Whether similar mechanisms account for fat reduction of C. elegans deficient in desaturase activity is not known. However, as in mammals, C. elegans delta-9 fatty acid desaturases are transcriptionally regulated by sterol response element binding protein and nuclear hormone receptors (see sections 4.1 and 4.2 below; Brock et al., 2006; Taubert et al., 2006; Van Gilst et al., 2005)
|
Figure 5. Coordination of fat synthesis and breakdown pathways by malonyl-CoA. AcetylCoA is the building block of fatty acids that are assembled into storage triglycerides through step-wise enzymatic processes. Inhibiting delta-9 desaturase activity (SCD-1 in mammals, fat-5, fat-6, and fat-7 in C. elegans) causes accumulation of saturated fatty acids. Consequently, acetylCoA carboxylase (ACC) is inactivated through feedback inhibition. This results in reduced levels of the ACC product malonyl-CoA. Since malonyl-CoA is an inhibitor of carnitine-palmitoyl-transferase (CPT), ACC inhibition results in activation of CPT, allowing uptake of fatty acids into mitochondria where they are broken down via β-oxidation. FAS: fatty acid synthase.
The capacity to coordinately adjust energy flux through various catabolic and anabolic pathways in response to changing nutritional status is critical for cellular and organismal survival. Metabolic sensing mechanisms are thought to coordinate these responses (Lindsley and Rutter, 2004). How alterations in energy status are sensed is a vibrant field of research. On a cellular level, metabolic sensors respond to altered concentrations of macronutrients, e.g., glucose, amino acids and fatty acids, metabolites derived from these macronutrients and energy resources such as ATP and NADH. In multicellular organisms, energetic status of different tissues is further coordinated through hormonal signals (Lindsley and Rutter, 2004; Salway, 2004). Recent studies in mammals indicate that some of these cellular metabolic sensors also function in the nervous system to regulate behavioral responses (Minokoshi et al., 2004; Porte et al., 2005). Several such C. elegans pathways are highlighted below:
Sterol response element binding protein (SREBP) is a key transcriptional regulator of fat and sterol synthesis pathways in mammals (Eberle et al., 2004; Rawson, 2003). RNAi inhibition and loss of function mutations in C. elegans SREBP, Y47D38.7/sbp-1/lpd-1, cause dramatic reductions in fat content and biogenesis of intestinal lipid droplets (Ashrafi, 2006; McKay et al., 2003; Yang et al., 2006). Thus far, analysis of candidate sbp-1 targets in C. elegans has been reported for malic enzyme (ME), and ATP citrate-lyase (ACL), acyl-CoA carboxylase (ACC), fatty acid synthase (FAS), stearoyl-CoA desaturases (FAT-6/FAT-7), and glycerol 3-phosphate acyltransferase (G3PA; Figure 5; McKay et al., 2003; Yang et al., 2006). The mammalian homologs of each of these lipogenic genes are direct SREBP targets. Additionally, sbp-1 regulates expression of elo-5 and elo-6, two fatty acid elongation enzymes required for synthesis of monomethyl branched chain fatty acids (Kniazeva et al., 2004).
Further conservation of function for sbp-1 has emerged from studies in which sbp-1 stimulated transcription of mammalian SREBP targets in a human cell line. Moreover, in both mammalian cells and C. elegans, SREBP function requires physical interaction with a transcriptional co-activator, ARC105/MDT-15 (Yang et al., 2006; see section 4.2).
Given that C. elegans are cholesterol auxotrophs, it remains to be determined whether any sterol metabolic pathways are regulated by sbp-1. One function of cholesterol in mammalian cells is to regulate membrane fluidity. Interestingly, Drosophila melanogaster, also a cholesterol auxotroph, regulates its membrane fluidity through an SREBP-mediated transcriptional program that produces phosphotidylethanolamine (Seegmiller et al., 2002). It is likely that a similar mechanism regulates membrane fluidity in C. elegans. Also, it remains to be determined if, as in mammals, C. elegans insulin signaling regulates sbp-1 function.
In mammals, several nuclear hormone receptors (NHRs) function as metabolic sensors and master regulators of energy balance (Chawla et al., 2001; Evans et al., 2004; Nakamura et al., 2004). The C. elegans genome contains a remarkable number of NHRs yet none display sequence homology to the Peroxisome Proliferator-Activated Receptor (PPAR) family, which are key regulators of fat, cholesterol and glucose homeostasis (Gissendanner et al., 2004; Sluder and Maina, 2001). Instead, the C. elegans NHR-49/MDT-15 system serves a parallel function to mammalian PPARα and its co-activator, PGC-1.
A deletion mutation in the HNF-4α family member nhr-49 mimics the high fat phenotype of nhr-49 RNAi (Ashrafi et al., 2003; Van Gilst et al., 2005). Analysis of genes in fat metabolic pathways (Table 1) by quantitative RT-PCR revealed that nhr-49 causes down-regulation of three genes encoding mitochondrial β-oxidation enzymes and concomitant up-regulation of three genes that encode peroxisomal β-oxidation enzymes (Van Gilst et al., 2005). Down-regulation of acs-2 and ech-1, two of the three affected mitochondrial β-oxidation genes, causes fat accumulation, and over-expression of acs-2 suppresses the high fat phenotype of nhr-49 (Van Gilst et al., 2005). This suggests that down-regulation of mitochondrial β-oxidation underlies excess fat levels of nhr-49 inactivation. Similar to mammalian PPARα nhr-49 also regulates expression of fatty acid desaturation and lipid binding proteins. Interestingly, changes in expression patterns of fat metabolic genes caused by nhr-49 inactivation overlap with expression changes noted after 12 hours of food deprivation (Van Gilst et al., 2005; Van Gilst et al., 2005). Thus, nhr-49 responds to nutrient signals and functions as a regulatory node of metabolic gene expression.
The transcriptional co-activator, mdt-15/arc105, was found to interact with nhr-49 in a yeast two-hybrid assay (Taubert et al., 2006). Many of the metabolic gene expression changes noted for nhr-49 are similarly altered in mdt-15/arc105 knock-down animals. Although overlapping, the full complement of expression changes caused by mdt-15/arc105 and nhr-49 are not identical, raising the possibility that mdt-15/arc105 functions as a co-activator for other transcription factors. Indeed, several other NHRs bind MDT-15/ARC105 in yeast two-hybrid experiments (Taubert et al., 2006). Interestingly, MDT15/ARC105 was recently characterized as a co-activator of SBP-1/SREBP in C. elegans and mammals (Yang et al., 2006). mdt-15/arc105, sbp-1, nhr-49, and nhr-80 control expression of delta-9 fatty acid desaturase genes (fat-5, fat-6, and fat-7) illustrating complex regulatory mechanisms of fat metabolic pathways (Brock et al., 2006; Taubert et al., 2006; Van Gilst et al., 2005).
Several other NHRs, whose mechanisms of function are unknown, are also required for wild type intestinal fat deposits (Ashrafi et al., 2003). Finally, another NHR, daf-12, and its co-factor din-1 are components of a steroid-based hormonal signaling pathway that controls entry into the high fat dauer state; however, the relationship of the DAF-12/DIN-1 complex to metabolic pathways is not yet clear (Antebi et al., 2000; Ludewig et al., 2004).
TOR (target of rapamyacin) is an evolutionarily conserved phosphatidylinositol kinase related family member that couples cell size and proliferation to nutrient levels, particularly amino acids and hormonal signals such as insulin (Inoki and Guan, 2006; Lindsley and Rutter, 2004). In Saccharomyces cerevisiae, Drosopholia melanogaster and mammalian cells, TOR activity promotes translation through direct activation of translational machinery. The precise nature of the nutrient signal that elicits TOR activity remains elusive. Loss of function mutations as well as RNAi inactivation of C. elegans TOR (let-363/B0261.2) and its partner Raptor (daf-15/C10C5.6) cause developmental arrest and fat accumulation (Jia et al., 2004; Vellai et al., 2003). Genetic analysis places raptor/daf-15 downstream of insulin signaling and upstream of the eIF-4G and eIF-2 subunits of translational machinery (Jia et al., 2004). In Drosophila melanogaster and mammals, the TSC tumor suppressor complex provides a mechanism of cross talk between the insulin and TOR signaling pathways. The C. elegans genome lacks sequence identifiable TSC complex components, TSC-1 and TSC-2. Thus, despite differences in cross talk mechanisms, insulin and TOR pathways interact in C. elegans to couple nutrient availability to growth and metabolism (Long et al., 2004; Long et al., 2002).
The AMP-activated kinase (AMPK) is a major cellular fuel gauge as its activity is responsive to cellular AMP:ATP ratio as well as upstream kinase cascades (Kahn et al., 2005; Lindsley and Rutter, 2004). AMPK activation causes numerous cellular changes, that together down-regulate energy-consumptive pathways and up-regulate energy-generating pathways. There is extensive cross talk between insulin, AMPK and TOR signaling pathways. In mammals, neuronal AMPK also functions downstream of leptin and insulin signaling to modulate food intake (Kahn et al., 2005; Lindsley and Rutter, 2004). Thus, AMPK is a major target of therapeutic intervention for metabolic syndromes such as type II diabetes.
Genes corresponding to catalytic α and regulatory β and γ subunits of AMPK are conserved in C. elegans and kinase activity for one of the two catalytic α subunits has been demonstrated (Apfeld et al., 2004; Curtis et al., 2006; Narbonne and Roy, 2006). Genetic analysis has linked the C. elegans AMPK cascade to insulin and mitochondrial pathways. These studies have focused on adult lifespan as a read-out. Direct investigation of the effect of C. elegans AMPK on fat regulatory pathways has not yet been reported.
O-linked N-acetylglucosamine (O-GlcNAc) is thought to function as a dynamic posttranslational modification of many proteins (Lindsley and Rutter, 2004; Love and Hanover, 2005). Production of uridine 5′-diphospho-N-GlcNAc (UDP-GlcNAc), the O-GlcNAc donor, occurs through the hexosamine biosynthetic pathway. O-GlcNAC transferase (OGT) and O-GlcNAC glycosidase (OGA) add and remove O-GlcNAc from target proteins. Flux through the hexosamine pathway is tuned to cellular energy levels. This allows for global alterations in functions of O-GlcNAC modified target proteins. Defects in this pathway are associated with numerous diseases including Type II diabetes. Deletions in C. elegans ogt-1 and oga-1 have been reported to increase glycogen and trehalose levels while decreasing fat levels. Moreover, insulin mediated dauer pathways are affected in ogt-1 and oga-1 mutant animals (Forsythe et al., 2006; Hanover et al., 2005; Love and Hanover, 2005).
During mammalian adipogenesis, hormonal cues initiate transcriptional programs that guide the differentiation of multipotent mesenchymal stem cells into mature adipocytes. Members of bZIP CCAAT/enhancer binding protein (C/EBP) transcription factor family and PPARγ are key components of these transcriptional cascades (Rosen, 2005). SREBP is also required for lipogenic programs of differentiating adipocytes. Despite the fact that C. elegans intestinal cells, the major site of fat deposition, are endodermal derivatives, the C. elegans counterpart of C/EBP (C48E7.11) and sbp-1 are expressed in intestinal cells and are required for fat storage (McKay et al., 2003). Electron microscopic examination of a deletion mutation of sbp-1, lpd-1(gf1), revealed that, intestinal cells of these animals maintain overall normal ultrastructural appearance including intact microvilli (McKay et al., 2003). This suggests that fat storage capacity of intestinal cells is distinct from developmental program of these cells as enterocytes.
McKay and colleagues also found that RNAi inactivation of each of eight genes (lpd-3 though lpd-9 and mac-1) causes morphological and fat phenotypes reminiscent of sbp-1 and C/EBP inactivation. lpd-4 (F26E9.4) and lpd-5 (ZK973.10) encode components of complex IV and complex I of the mitochondrial respiratory chain, respectively. Chemical inhibition of complex I and complex IV by rotenone and NaN3 causes reduction of lipid accumulation in 3T3-L1 cells, a murine tissue culture adipocyte model system. Moreover, lpd-3 encodes a novel but conserved gene expressed in C. elegans intestinal cells. The mammalian counterpart of lpd-3 is strongly expressed in brain, testis and embryonic fat tissues. Inactivation of mammalian lpd-3 by shRNA in 3T3-L1 cells that had been induced to undergo adipogenesis prevented lipid accumulation despite appearance of adipocyte differentiation markers (McKay et al., 2003).
The C. elegans genome encodes proteins with sequence homology to fatty acid translocase (FAT/CD36), fatty acid transport protein (FATP), fatty acid binding proteins (FABPs), acyl-CoA binding proteins (ACBPs), carnitine-palmitoyl transferases (CPTs) and ATP-binding-cassette (ABC) transporter proteins. Mammalian homologs of these genes mediate fatty acid transport across various lipid bilayers and intracellular shuttling of fatty acylCoAs. In mammals, altered expression of putative fatty acid transport proteins is associated with obesity and insulin resistant states (Chawla et al., 2001; Koonen et al., 2005; Mashek and Coleman, 2006).
Loss of function mutations and RNAi inactivation of specific members of FABP, ACBP, CPT and ABC transporters cause increased or decreased intestinal fat levels as visualized by Nile Red staining (Ashrafi et al., 2003). Loss of function of a FATP-like transporter causes a reduced fat phenotype only in the context of mutations that confer increased fat storage (K. Ashrafi, unpublished results). Additionally, RNAi inactivations of specific family members of OCT-type transporters, a lysosomal transporter, an amino-acid permease and glucose transporters cause altered fat accumulation (Ashrafi et al., 2003). Mechanisms of function underlying these phenotypes are not known.
Similarly, inactivation of the opt-2/pep-2 and nhx-2 transporters cause reduced fat accumulation (Ashrafi et al., 2003; Meissner et al., 2004; Nehrke, 2003). GFP-reporter fusions for each of these genes are exclusively expressed along the apical membrane of intestinal epithelia. OPT-2 is a transporter of di- and tripeptides and NHX-2 is a Na+/H+ exchanger. These functions are required for appropriate acidification of intestinal cells, which in turn, powers a variety of proton-coupled nutrient uptake systems.
The best characterized lipid uptake and transport system in C. elegans has been delivery of nutrients from intestinal cells to developing embryos (see Intracellular trafficking). A mixture of fats and cholesterol are loaded onto vitellogenins, yolk proteins with functional and structural similarities to LDL-type proteins. Vitellogenins are secreted from the intestinal cells into the pseudocoelum and then taken up by developing embryos via receptor-mediated endocytosis (Fares and Grant, 2002). Finally, a conserved, transmembrane ACBP, maa-1, is associated with Golgi and endosomal membranes. This ACBP modulates vesicular transport in the intestine, hypodermis and oocytes and, when inactivated, impairs receptor-mediated endocytosis (Larsen et al., 2006).
In mammals, the nervous system functions as a central coordinator of both metabolic pathways and behaviors associated with food consumption. The C. elegans nervous system also regulates fat storage both in conjunction with and independent of feeding pathways.
Signaling cascades through insulin, transforming growth factor (TGF-β) and cyclic nucleotide regulated pathways control whether C. elegans larvae grow to adults or fat-storing dauers. Molecular components of these pathways are extensively covered elsewhere. Down-regulating either insulin or TGF-β pathway components promotes fat accumulation in adults. A clear example of neuronal regulation of fat levels is provided by DAF-7, a TGF-β ligand. daf-7 is expressed in one pair of ciliated sensory neurons (ASI) and its transcription is modulated by daumone, a constitutively secreted pheromone that C. elegans use to assess population density (Jeong et al., 2005; Ren et al., 1996). Thus, this pathway responds to environmental conditions and function as a central regulator of C. elegans homeostasis.
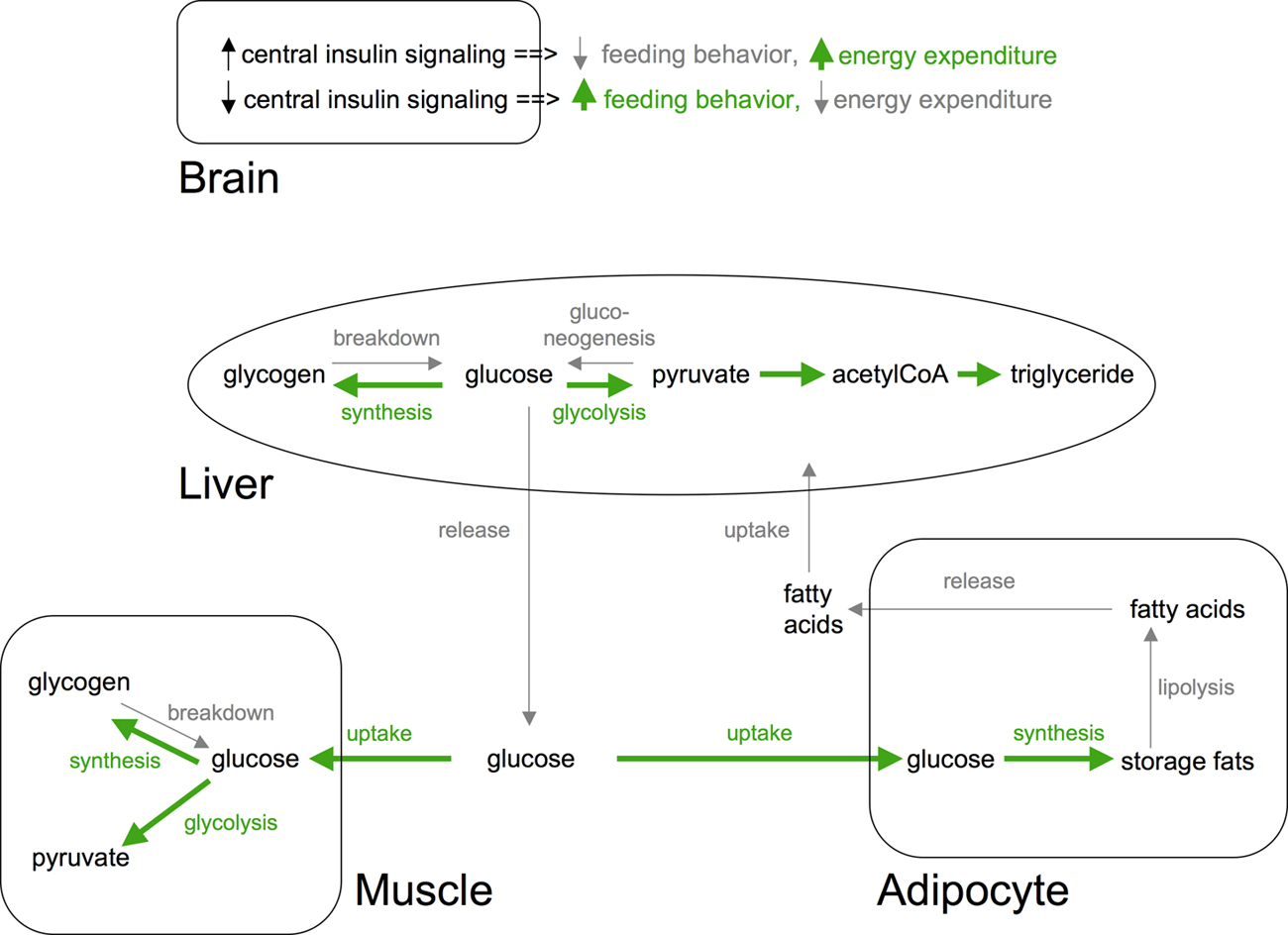
Similarly, many of the C. elegans insulins are expressed in neurons and have been postulated to relay changes in food availability, although direct evidence for this is lacking (Pierce et al., 2001). In mammals, insulin signaling has both peripheral and central actions on fat homeostasis (see Figure 6). Importantly, tissue-specific knockouts or reconstitution of the insulin receptor in mice has begun to reveal contributions of different tissues to glucose and fat homeostasis. For instance, neuronal insulin receptor knockout and muscle insulin receptor knockout mice are obese while fat cell insulin receptor knockout mice are lean and resistant to diet induced obesity (Biddinger and Kahn, 2006). Similarly, insulin signaling in different C. elegans tissues contributes differentially to fat content. For instance, reconstitution of the insulin receptor in neurons but not in muscle partially rescues the increased fat content of insulin receptor knockout animals (Wolkow et al., 2000).
|
Figure 6. Systemic actions of insulin signaling in mammals. In response to nutrients (e.g., glucose) insulin signaling promotes uptake and storage of glucose as glycogen and triglycerides in muscle and adipocytes, respectively. Concomitantly, insulin inhibits triglyceride and glycogen breakdown pathways in these tissues. Similarly, insulin inhibits hepatic gluconeogenesis and glycongenolysis while promoting glycogen synthesis. In both liver and muscle, insulin also promotes glucose breakdown. Insulin signaling in brain inhibits feeding behavior and activates energy expenditure pathways. Tissue-specific insulin receptor knockouts have revealed complex compensatory mechanisms. Pathways activated by insulin signaling are shown in green. Pathways inhibited are shown in light gray. Additional actions of insulin on protein synthesis/breakdown and other tissues are not shown.
Classical neurotransmitters have dramatic effects on fat regulation in nemotodes and in mammals. Deleting tph-1, which encodes the enzyme tryptophan hydroxylase, causes C. elegans to lack serotonin (Sze et al., 2000). Serotonin production is largely confined to ADF, a head sensory neuron, NSM, a pharyngeal neuron, HSN, a hermaphrodite-specific neuron that innervates the vulva, and sensory neurons innervating the male tail. Serotonin-deficient animals are viable but have excess fat accumulation, reduced feeding rate (see section 7.4) and reduced rate of progeny production (Sze et al., 2000). Genetic analysis suggests interactions between serotonin, insulin and TGF-β pathways (Sze et al., 2000).
Additionally, inactivating specific dopaminergic and glutamergic receptors alters fat deposits without adversely affecting growth rate or viability (Ashrafi et al., 2003). Mammalian counterparts of these neuromodulatory pathways have also been implicated energy balance (Clifton and Kennett, 2006; Sainsbury et al., 2002).
Mutations in rodent tubby cause progressive degeneration in retinal and cochlear sensory receptor cells, infertility and adult-onset obesity with insulin resistance (Carroll et al., 2004). Tubby is broadly expressed in the central nervous system including the hypothalamus. Molecular mechanisms of Tubby function are unclear although several models have been proposed (Carroll et al., 2004). Loss of function in tub-1/F10B5.5, the C. elegans ortholog of Tubby, causes fat accumulation (Ashrafi et al., 2003; Mak et al., 2006). A functional TUB-1::GFP fusion localizes to all ciliated sensory neurons in C. elegans (Mak et al., 2006; Mukhopadhyay et al., 2005). In a yeast two-hybrid assay, TUB-1 was found to interact with B0393.2, a predicted RabGTPase-activating protein (RabGAP) postulated to function in vesicular transport (Mukhopadhyay et al., 2005). This RabGAP is expressed in the amphid and phasmid subset of ciliated sensory neurons. RNAi inactivation of this RabGAP causes only a minor reduction in fat content of wild-type animals but suppresses the excess fat of tub-1 mutant animals (Mukhopadhyay et al., 2005). This suggests a surprisingly specific role for vesicular transport in accumulation of excess fat in tub-1 deficient animals. Moreover, tub-1 mutant animals have extended lifespan. This lifespan extension requires insulin signaling but appears to be independent of the TUB-RabGAP fat pathway (Mukhopadhyay et al., 2005).
Neuronal tub-1 has been reported to act synergistically in fat accumulation with kat-1/T02G5.8, which encodes a non-neuronal β-oxidation enzyme, 3-ketoacyl-coA thiolase (Mak et al., 2006). The synergistic nature of the excess fat accumulation in tub-1;kat-1 double mutants suggests that defects in neuronal tub-1 are normally compensated by kat-1 mediated fat oxidation in non-neuronal tissues. Loss of kat-1 abrogates this multi-tissue compensatory mechanism.
The molecular nature of compensatory mechanisms that couple tub-1 and kat-1 are not yet known; however, genetic analysis of kat-1 led to identification of bbs-1 as another modifier of intestinal fat storage that, like tub-1, functions in ciliated neurons (Mak et al., 2006). Mutations in human ortholog of bbs genes including bbs-1 underlie Bardet-Biedl syndrome, a pleiotropic syndrome associated with obesity (Beales, 2005). Many human BBS genes, which are implicated in ciliogenesis and intraflagellar transport (IFT), have C. elegans homologs (Inglis et al., 2006). Similar to tub-1, loss of function mutations in bbs-1 cause modest increases in fat accumulation that are exacerbated by loss of KAT-1 (Mak et al., 2006; Mukhopadhyay et al., 2005). Moreover, tub-1 mutants have defects in chemotaxis, a function mediated by a subset of ciliated sensory neurons, and there is evidence that TUB-1 undergoes IFT (Mak et al., 2006; Mukhopadhyay et al., 2005; see Chemosensation). Together, these findings suggest that tub-1 and bbs-1 function in the same fat regulatory pathway.
The provocative hypothesis that bbs-1 and tub-1 form a neuroendocrine axis with kat-1 is based on the synergistic rather than additive fat content of double mutants as assessed by Nile Red fluorescence. The potential insights offered by such genetic interactions highlight the need for standard methods to accurately quantify fluorescence intensity.
C. elegans feed by pumping and concentrating food using a neuromuscular organ known as the pharynx (Avery and Shtonda, 2003; Shtonda and Avery, 2005). The grinder, a teeth-like structure located at the junction of the pharynx and the intestine, breaks food particles that are then pushed into the lumen of the intestine by the peristaltic pumping action of the pharynx. C. elegans pump in the presence and absence of food; however, pumping rate is modulated by food availability (Avery and Horvitz, 1990). Animals that have experienced starvation will pump faster when re-exposed to food than well-fed animals. C. elegans also forage for food. Rates and patterns of C. elegans movement are different compared on or off food. These locomotory rates and patterns are also modulated by starvation (Hills et al., 2004; Sawin et al., 2000).
Serotonin modulates pumping rate. tph-1 mutant animals display reduced pumping rate while animals exposed to excess serotonin or imipramine, a serotonin uptake inhibitor, display increased pumping (Avery and Horvitz, 1990; Horvitz et al., 1982). Pumping stimulatory effects of serotonin are abrogated by mutations in each of two serotonergic receptors ser-1 and ser-7 (Hobson et al., 2006). Additionally, serotonin, dopamine and glutamate signaling pathways are implicated in different foraging strategies of C. elegans (Hills et al., 2004; Sawin et al., 2000). These neuronal signaling mechanisms also modulate mammalian feeding behavior (Clifton and Kennett, 2006; Sainsbury et al., 2002).
Together, these results indicate an overlap between neuronal feeding and foraging behavior pathways and central fat regulatory mechanisms; however, the nature of these relationships is not yet clear. For instance, there is an inverse correlation between fat content and pumping rate for serotonin deficient animals. In other cases, such as tub-1 mutants, animals display wild-type pumping rates despite increased fat levels.
Our understanding of body fat regulation as a homeostatic, organismal process has flourished in the past decade. Although many of the core metabolic pathways were biochemically defined long ago, integration and coordination of these pathways across multiple tissues is a vibrant field of integrative biology. This is because understanding fat regulation requires multiple layers of investigation spanning from metabolism, transcription and signaling to neuronal development and behavior. Deciphering neuronal circuits that coordinate behavior, physiology, and metabolism is a major challenge in understanding fat regulation. Similarly, compensatory mechanisms that operate at organismal level to maintain energy homeostasis are just being elucidated.
The genetic tractability of C. elegans has already revealed that mechanisms of energy balance in this organism range from neuronal sensation and endocrine signals to nutrient uptake, transport and storage/utilization mechanisms. Importantly, amenability of C. elegans to multiple rounds of suppressor/enhancer screening is critical and provides a unique advantage for understanding homeostatic feedback regulatory mechanisms. Examining fat regulatory pathways under different environmental conditions holds the potential to reveal how physiological pathways are coordinately modulated in response to environmental perturbation. Similarly, how developmental stage, age, experience and diet perturb and possibly rewire the fat networks can be addressed in C. elegans at a molecular level. Finally, C. elegans is well suited for deciphering developmental programs that underlie fat storage capacity and cell biological determinants of lipid droplet biogenesis.
Many of the adverse health effects of excess fat accumulation in humans are unlikely to occur in C. elegans. Nevertheless, the limited number of studies reported thus far already reveal remarkable similarities between molecular components of mammalian and C. elegans fat pathways that extend to disease-associated genes. Many of the fat genes identified in C. elegans have mammalian homologs whose roles in energy balance have not yet been examined. Given that energy balance is fundamental for viability, it is likely that many of the newly identified C. elegans fat regulatory networks are functionally conserved in mammals.
Work in the Ashrafi laboratory is supported by a Career Award in Biomedical Sciences from Burroughs Welcome Fund, and grants from the NIH, Searle Scholars Program, ADA/Smith Family Foundation and The UCSF Sandler Program in Basic Sciences. I am indebted to members of the Ashrafi lab and Jennifer Watts for discussions.
Allison, D.B., Neale, M.C., Kezis, M.I., Alfonso, V.C., Heshka, S., and Heymsfield, S.B. (1996). Assortative mating for relative weight: genetic implications. Behav. Genet. 26, 103–111. Abstract
Antebi, A., Yeh, W.H., Tait, D., Hedgecock, E.M., and Riddle, D.L. (2000). daf-12 encodes a nuclear receptor that regulates the dauer diapause and developmental age in C. elegans. Genes Dev. 14, 1512–1527. Abstract Article
Apfeld, J., O'Connor, G., McDonagh, T., DiStefano, P.S., and Curtis, R. (2004). The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 18, 3004–3009. Abstract Article
Ashrafi, K., Chang, F.Y., Watts, J.L., Fraser, A.G., Kamath, R.S., Ahringer, J., and Ruvkun, G. (2003). Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 421, 268–272. Abstract Article
Avery, L., and Horvitz, H.R. (1990). Effects of starvation and neuroactive drugs on feeding in Caenorhabditis elegans. J. Exp. Zool. 253, 263–270. Abstract Article
Avery, L., and Shtonda, B.B. (2003). Food transport in the C. elegans pharynx. J. Exp. Biol. 206, 2441–2457. Abstract Article
Badman, M.K., and Flier, J.S. (2005). The gut and energy balance: visceral allies in the obesity wars. Science 307, 1909–1914. Abstract Article
Barsh, G.S., Farooqi, I.S., and O'Rahilly, S. (2000). Genetics of body-weight regulation. Nature 404, 644–651. Abstract Article
Beales, P.L. (2005). Lifting the lid on Pandora's box: the Bardet-Biedl syndrome. Curr. Opin. Genet. Dev. 15, 315–323. Abstract Article
Biddinger, S.B., and Kahn, C.R. (2006). From mice to men: insights into the insulin resistance syndromes. Annu. Rev. Physiol. 68, 123–158. Abstract Article
Braeckman, B.P., Houthoofd, K., and Vanfleteren, J.R. (2002). Assessing metabolic activity in aging Caenorhabditis elegans: concepts and controversies. Aging Cell 1, 82–88; discussion 102–103. Abstract Article
Brock, T.J., Browse, J., and Watts, J.L. (2006). Genetic regulation of unsaturated fatty acid composition in C. elegans. PLoS Genet. 2, e108. Abstract Article
Brockmann, G.A., and Bevova, M.R. (2002). Using mouse models to dissect the genetics of obesity. Trends Genet. 18, 367–376. Abstract
Burnell, A.M., Houthoofd, K., O'Hanlon, K., and Vanfleteren, J.R. (2005). Alternate metabolism during the dauer stage of the nematode Caenorhabditis elegans. Exp. Gerontol. 40, 850–856. Abstract Article
Carroll, K., Gomez, C., and Shapiro, L. (2004). Tubby proteins: the plot thickens. Nat. Rev. Mol. Cell Biol. 5, 55–63. Abstract Article
Chawla, A., Repa, J.J., Evans, R.M., and Mangelsdorf, D.J. (2001). Nuclear receptors and lipid physiology: opening the X-files. Science 294, 1866–1870. Abstract Article
Clifton, P.G., and Kennett, G.A. (2006). Monoamine receptors in the regulation of feeding behaviour and energy balance. CNS Neurol. Disord. Drug Targets 5, 293–312. Abstract Article
Cohen, P., and Friedman, J.M. (2004). Leptin and the control of metabolism: role for stearoyl-CoA desaturase-1 (SCD-1). J. Nutr. 134, 2455S–2463S. Abstract
Cohen, P., Ntambi, J.M., and Friedman, J.M. (2003). Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr. Drug Targets Immune Endocr. Metabol. Disord. 3, 271–280. Abstract
Cone, R.D. (2005). Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 8, 571–578. Abstract Article
Curtis, R., O'Connor, G., and DiStefano, P.S. (2006). Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell 5, 119–126. Abstract Article
Delrue, M.A., and Michaud, J.L. (2004). Fat chance: genetic syndromes with obesity. Clin. Genet. 66, 83–93. Abstract Article
Demuro, G., and Obici, S. (2006). Central nervous system and control of endogenous glucose production. Curr. Diab. Rep. 6, 188–193. Abstract Article
Eberle, D., Hegarty, B., Bossard, P., Ferre, P., and Foufelle, F. (2004). SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848. Abstract Article
Evans, R.M., Barish, G.D., and Wang, Y.X. (2004). PPARs and the complex journey to obesity. Nat. Med. 10, 355–361. Abstract Article
Fares, H., and Grant, B. (2002). Deciphering endocytosis in Caenorhabditis elegans. Traffic 3, 11–19. Abstract Article
Flier, J.S. (2004). Obesity wars: molecular progress confronts an expanding epidemic. Cell 116, 337–350. Abstract Article
Forsythe, M.E., Love, D.C., Lazarus, B.D., Kim, E.J., Prinz, W.A., Ashwell, G., Krause, M.W., and Hanover, J.A. (2006). Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer. Proc. Natl. Acad. Sci. U.S.A. 103, 11952–11957. Abstract Article
Friedman, J.M. (2002). The function of leptin in nutrition, weight, and physiology. Nutr. Rev. 60, S1–14; discussion S68–84, 85–17. Abstract Article
Friedman, J.M., and Halaas, J.L. (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763–770. Abstract Article
Gems, D. (1999). Nematode ageing: Putting metabolic theories to the test. Curr. Biol. 9, R614–R616. Abstract Article
Gissendanner, C.R., Crossgrove, K., Kraus, K.A., Maina, C.V., and Sluder, A.E. (2004). Expression and function of conserved nuclear receptor genes in Caenorhabditis elegans. Dev. Biol. 266, 399–416. Abstract Article
Halaschek-Wiener, J., Khattra, J.S., McKay, S., Pouzyrev, A., Stott, J.M., Yang, G.S., Holt, R.A., Jones, S.J., Marra, M.A., Brooks-Wilson, A.R., and Riddle, D.L. (2005). Analysis of long-lived C. elegans daf-2 mutants using serial analysis of gene expression. Genome Res. 15, 603–615. Abstract Article
Hanover, J.A., Forsythe, M.E., Hennessey, P.T., Brodigan, T.M., Love, D.C., Ashwell, G., and Krause, M. (2005). A Caenorhabditis elegans model of insulin resistance: altered macronutrient storage and dauer formation in an OGT-1 knockout. Proc. Natl. Acad. Sci. U.S.A. 102, 11266–11271. Abstract Article
Hills, T., Brockie, P.J., and Maricq, A.V. (2004). Dopamine and glutamate control area-restricted search behavior in Caenorhabditis elegans. J. Neurosci. 24, 1217–1225. Abstract Article
Hobson, R.J., Hapiak, V.M., Xiao, H., Buehrer, K.L., Komuniecki, P.R., and Komuniecki, R.W. (2006). SER-7, a Caenorhabditis elegans 5-HT7-like receptor, is essential for the 5-HT stimulation of pharyngeal pumping and egg laying. Genetics 172, 159–169. Abstract Article
Holt, S.J., and Riddle, D.L. (2003). SAGE surveys C. elegans carbohydrate metabolism: evidence for an anaerobic shift in the long-lived dauer larva. Mech. Ageing Dev. 124, 779–800. Abstract Article
Horvitz, H.R., Chalfie, M., Trent, C., Sulston, J.E., and Evans, P.D. (1982). Serotonin and octopamine in the nematode Caenorhabditis elegans. Science 216, 1012–1014. Abstract Article
Inglis, P.N., Boroevich, K.A., and Leroux, M.R. (2006). Piecing together a ciliome. Trends Genet. 22, 491–500. Abstract Article
Inoki, K., and Guan, K.L. (2006). Complexity of the TOR signaling network. Trends Cell Biol. 16, 206–212. Abstract Article
Jeong, P.Y., Jung, M., Yim, Y.H., Kim, H., Park, M., Hong, E., Lee, W., Kim, Y.H., Kim, K., and Paik, Y.K. (2005). Chemical structure and biological activity of the Caenorhabditis elegans dauer-inducing pheromone. Nature 433, 541–545. Abstract Article
Jia, K., Chen, D., and Riddle, D.L. (2004). The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131, 3897–3906. Abstract Article
Jones, S.J., Riddle, D.L., Pouzyrev, A.T., Velculescu, V.E., Hillier, L., Eddy, S.R., Stricklin, S.L., Baillie, D.L., Waterston, R., and Marra, M.A. (2001). Changes in gene expression associated with developmental arrest and longevity in Caenorhabditis elegans. Genome Res. 11, 1346–1352. Abstract Article
Kahn, B.B., Alquier, T., Carling, D., and Hardie, D.G. (2005). AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25. Abstract Article
Kahn-Kirby, A.H., Dantzker, J.L., Apicella, A.J., Schafer, W.R., Browse, J., Bargmann, C.I., and Watts, J.L. (2004). Specific polyunsaturated fatty acids drive TRPV-dependent sensory signaling in vivo. Cell 119, 889–900. Abstract Article
Kimura, K.D., Tissenbaum, H.A., Liu, Y., and Ruvkun, G. (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–946. Abstract
Kniazeva, M., Crawford, Q.T., Seiber, M., Wang, C.Y., and Han, M. (2004). Monomethyl branched-chain fatty acids play an essential role in Caenorhabditis elegans development. PLoS Biol. 2, E257. Abstract Article
Kniazeva, M., Sieber, M., McCauley, S., Zhang, K., Watts, J.L., and Han, M. (2003). Suppression of the ELO-2 FA elongation activity results in alterations of the fatty acid composition and multiple physiological defects, including abnormal ultradian rhythms, in Caenorhabditis elegans. Genetics 163, 159–169. Abstract Article
Koonen, D.P., Glatz, J.F., Bonen, A., and Luiken, J.J. (2005). Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim. Biophys. Acta 1736, 163–180. Abstract Article
Kurzchalia, T.V., and Ward, S. (2003). Why do worms need cholesterol? Nat. Cell Biol. 5, 684–688. Abstract Article
Lam, T.K., Schwartz, G.J., and Rossetti, L. (2005). Hypothalamic sensing of fatty acids. Nat. Neurosci. 8, 579–584. Abstract Article
Larsen, M.K., Tuck, S., Faergeman, N.J., and Knudsen, J. (2006). MAA-1, a Novel Acyl-CoA-binding Protein Involved in Endosomal Vesicle Transport in Caenorhabditis elegans. Mol. Biol. Cell. Abstract Article
Larsen, P.L., Albert, P.S., and Riddle, D.L. (1995). Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics 139, 1567–1583. Abstract
Lee, S.S., Kennedy, S., Tolonen, A.C., and Ruvkun, G. (2003). DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644–647. Abstract Article
Lesa, G.M., Palfreyman, M., Hall, D.H., Clandinin, M.T., Rudolph, C., Jorgensen, E.M., and Schiavo, G. (2003). Long chain polyunsaturated fatty acids are required for efficient neurotransmission in C. elegans. J. Cell. Sci. 116, 4965–4975. Abstract Article
Lindsley, J.E., and Rutter, J. (2004). Nutrient sensing and metabolic decisions. Comp. Biochem. Physiol. B, Biochem. Mol. Biol. 139, 543–559. Abstract Article
Long, X., Muller, F., and Avruch, J. (2004). TOR action in mammalian cells and in Caenorhabditis elegans. Curr. Top. Microbiol. Immunol. 279, 115–138. Abstract
Long, X., Spycher, C., Han, Z.S., Rose, A.M., Muller, F., and Avruch, J. (2002). TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr. Biol. 12, 1448–1461. Abstract Article
Love, D.C., and Hanover, J.A. (2005). The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci. STKE 2005, re13. Abstract Article
Luchsinger, J.A. (2006). A work in progress: the metabolic syndrome. Sci. Aging Knowledge Environ. 2006, pe19. Abstract Article
Ludewig, A.H., Kober-Eisermann, C., Weitzel, C., Bethke, A., Neubert, K., Gerisch, B., Hutter, H., and Antebi, A. (2004). A novel nuclear receptor/coregulator complex controls C. elegans lipid metabolism, larval development, and aging. Genes Dev. 18, 2120–2133. Abstract Article
Lund, J., Tedesco, P., Duke, K., Wang, J., Kim, S.K., and Johnson, T.E. (2002). Transcriptional profile of aging in C. elegans. Curr. Biol. 12, 1566–1573. Abstract Article
Mak, H.Y., Nelson, L.S., Basson, M., Johnson, C.D., and Ruvkun, G. (2006). Polygenic control of Caenorhabditis elegans fat storage. Nat. Genet. 38, 363–368. Abstract Article
Mashek, D.G., and Coleman, R.A. (2006). Cellular fatty acid uptake: the contribution of metabolism. Curr. Opin. Lipidol. 17, 274–278. Abstract Article
McElwee, J.J., Schuster, E., Blanc, E., Thomas, J.H., and Gems, D. (2004). Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 279, 44533–44543. Abstract Article
McElwee, J.J., Schuster, E., Blanc, E., Thornton, J., and Gems, D. (2006). Diapause-associated metabolic traits reiterated in long-lived daf-2 mutants in the nematode Caenorhabditis elegans. Mech. Ageing Dev. 127, 458–472. Abstract Article
McKay, R.M., McKay, J.P., Avery, L., and Graff, J.M. (2003). C. elegans: a model for exploring the genetics of fat storage. Dev. Cell 4, 131–142. Abstract Article
Meissner, B., Boll, M., Daniel, H., and Baumeister, R. (2004). Deletion of the intestinal peptide transporter affects insulin and TOR signaling in Caenorhabditis elegans. J. Biol. Chem. 279, 36739–36745. Abstract Article
Minokoshi, Y., Alquier, T., Furukawa, N., Kim, Y.B., Lee, A., Xue, B., Mu, J., Foufelle, F., Ferre, P., Birnbaum, M.J., et al. (2004). AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574. Abstract Article
Mukhopadhyay, A., Deplancke, B., Walhout, A.J., and Tissenbaum, H.A. (2005). C. elegans tubby regulates life span and fat storage by two independent mechanisms. Cell Metab. 2, 35–42. Abstract Article
Murphy, C.T., McCarroll, S.A., Bargmann, C.I., Fraser, A., Kamath, R.S., Ahringer, J., Li, H., and Kenyon, C. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283. Abstract Article
Nakamura, M.T., Cheon, Y., Li, Y., and Nara, T.Y. (2004). Mechanisms of regulation of gene expression by fatty acids. Lipids 39, 1077–1083. Abstract Article
Narbonne, P., and Roy, R. (2006). Inhibition of germline proliferation during C. elegans dauer development requires PTEN, LKB1 and AMPK signalling. Development 133, 611–619. Abstract Article
Nehrke, K. (2003). A reduction in intestinal cell pHi due to loss of the Caenorhabditis elegans Na+/H+ exchanger NHX-2 increases life span. J. Biol. Chem. 278, 44657–44666. Abstract Article
Nunes, F., Wolf, M., Hartmann, J., and Paul, R.J. (2005). The ABC transporter PGP-2 from Caenorhabditis elegans is expressed in the sensory neuron pair AWA and contributes to lysosome formation and lipid storage within the intestine. Biochem. Biophys. Res. Commun. 338, 862–871. Abstract Article
Pierce, S.B., Costa, M., Wisotzkey, R., Devadhar, S., Homburger, S.A., Buchman, A.R., Ferguson, K.C., Heller, J., Platt, D.M., Pasquinelli, A.A., et al. (2001). Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 15, 672–686. Abstract Article
Porte, D., Jr., Baskin, D.G., and Schwartz, M.W. (2002). Leptin and insulin action in the central nervous system. Nutr. Rev. 60, S20–S29; discussion S68–S84, 85–27. Abstract Article
Porte, D., Jr., Baskin, D.G., and Schwartz, M.W. (2005). Insulin signaling in the central nervous system: a critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes 54, 1264–1276. Abstract
Rawson, R.B. (2003). The SREBP pathway--insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 4, 631–640. Abstract Article
Ren, P., Lim, C.S., Johnsen, R., Albert, P.S., Pilgrim, D., and Riddle, D.L. (1996). Control of C. elegans larval development by neuronal expression of a TGF-beta homolog. Science 274, 1389–1391. Abstract Article
Rondinone, C.M. (2006). Adipocyte-derived hormones, cytokines, and mediators. Endocrine 29, 81–90. Abstract Article
Rosen, E.D. (2005). The transcriptional basis of adipocyte development. Prostaglandins Leukot. Essent. Fatty Acids 73, 31–34. Abstract Article
Sainsbury, A., Cooney, G.J., and Herzog, H. (2002). Hypothalamic regulation of energy homeostasis. Best Pract. Res. Clin. Endocrinol. Metab. 16, 623–637. Abstract Article
Satouchi, K., Hirano, K., Sakaguchi, M., Takehara, H., and Matsuura, F. (1993). Phospholipids from the free-living nematode Caenorhabditis elegans. Lipids 28, 837–840. Abstract Article
Sawin, E.R., Ranganathan, R., and Horvitz, H.R. (2000). C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26, 619–631. Abstract Article
Seegmiller, A.C., Dobrosotskaya, I., Goldstein, J.L., Ho, Y.K., Brown, M.S., and Rawson, R.B. (2002). The SREBP pathway in Drosophila: regulation by palmitate, not sterols. Dev. Cell 2, 229–238. Abstract Article
Shtonda, B., and Avery, L. (2005). CCA-1, EGL-19 and EXP-2 currents shape action potentials in the Caenorhabditis elegans pharynx. J. Exp. Biol. 208, 2177–2190. Abstract Article
Sluder, A.E., and Maina, C.V. (2001). Nuclear receptors in nematodes: themes and variations. Trends Genet. 17, 206–213. Abstract Article
Spiegelman, B.M., and Flier, J.S. (2001). Obesity and the regulation of energy balance. Cell 104, 531–543. Abstract Article
Stunkard, A.J., Harris, J.R., Pedersen, N.L., and McClearn, G.E. (1990). The body-mass index of twins who have been reared apart. N. Engl. J. Med. 322, 1483–1487. Abstract
Sze, J.Y., Victor, M., Loer, C., Shi, Y., and Ruvkun, G. (2000). Food and metabolic signalling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature 403, 560–564. Abstract Article
Tanaka, T., Ikita, K., Ashida, T., Motoyama, Y., Yamaguchi, Y., and Satouchi, K. (1996). Effects of growth temperature on the fatty acid composition of the free-living nematode Caenorhabditis elegans. Lipids 31, 1173–1178. Abstract Article
Taubert, S., Van Gilst, M.R., Hansen, M., and Yamamoto, K.R. (2006). A Mediator subunit, MDT-15, integrates regulation of fatty acid metabolism by NHR-49-dependent and -independent pathways in C. elegans. Genes Dev. 20, 1137–1149. Abstract Article
Trayhurn, P., and Bing, C. (2006). Appetite and energy balance signals from adipocytes. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 361, 1237–1249. Abstract Article
Van Gilst, M.R., Hadjivassiliou, H., Jolly, A., and Yamamoto, K.R. (2005). Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 3, e53. Abstract Article
Van Gilst, M.R., Hadjivassiliou, H., and Yamamoto, K.R. (2005). A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc. Natl. Acad. Sci. U.S.A. 102, 13496–13501. Abstract Article
Van Voorhies, W.A., and Ward, S. (1999). Genetic and environmental conditions that increase longevity in Caenorhabditis elegans decrease metabolic rate. Proc. Natl. Acad. Sci. U.S.A. 96, 11399–11403. Abstract Article
Vellai, T., Takacs-Vellai, K., Zhang, Y., Kovacs, A.L., Orosz, L., and Muller, F. (2003). Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426, 620. Abstract Article
Wadsworth, W.G., and Riddle, D.L. (1989). Developmental regulation of energy metabolism in Caenorhabditis elegans. Dev. Biol. 132, 167–173. Abstract Article
Wallis, J.G., Watts, J.L., and Browse, J. (2002). Polyunsaturated fatty acid synthesis: what will they think of next? Trends Biochem. Sci. 27, 467. Abstract Article
Wang, J., and Kim, S.K. (2003). Global analysis of dauer gene expression in Caenorhabditis elegans. Development 130, 1621–1634. Abstract Article
Watts, J.L., and Browse, J. (2002). Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 99, 5854–5859. Abstract Article
Watts, J.L., and Browse, J. (2006). Dietary manipulation implicates lipid signaling in the regulation of germ cell maintenance in C. elegans. Dev. Biol. 292, 381–392. Abstract Article
Watts, J.L., Phillips, E., Griffing, K.R., and Browse, J. (2003). Deficiencies in C20 polyunsaturated fatty acids cause behavioral and developmental defects in Caenorhabditis elegans fat-3 mutants. Genetics 163, 581–589. Abstract
*Edited by Andres Villu Maricq and Steven L. McIntire. Last revised November 30, 2006. Published March 9, 2007. This chapter should be cited as: Ashrafi, K. Obesity and the regulation of fat metabolism (March 9, 2007), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.130.1, http://www.wormbook.org.
Copyright: © 2007 Kaveh Ashrafi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Tel: 415-514-4102, Fax: 415-514-4242. E-mail: kaveh.ashrafi@ucsf.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.