Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard Schierenberg
Table of Contents
Caenorhabditis briggsae is being developed in parallel to C. elegans as a model system, primarily for the study of evolution. Like C. elegans, C. briggsae is a protandrous hermaphrodite (Nigon and Dougherty, 1949) and like C. elegans, its genome has been sequenced (Stein et al., 2003). From this point, these two model systems diverge. The development, behavior, and physiology of C. elegans have been characterized through tens of thousands of genetic and molecular studies. Genetic and molecular characterizations of C. briggsae are relatively few. Experimental resources in C. elegans include a high density recombination map that is well integrated with the genome sequence. The C. briggsae recombination map has yet to be published and attempts to integrate it with the genome sequence are in their infancy. Despite these deficiencies, C. briggsae is attractive for several reasons. First as a parallel system, it can be used to test the generality of results obtained in C. elegans. Second, it appears that the structure of the C. briggsae world-wide population is qualitatively different from that of C. elegans and that C. briggsae may be more amenable to studies of gene flow, genome evolution, and speciation (Cutter et al., 2006; Baird and Hampton, unpubl. results). Finally, C. briggsae and C. remanei are sister species with C. elegans as an outgroup, making the C. briggsae–C. remanei species better for some aspects of comparative genomics (Kiontke et al., 2004; Cho et al., 2004; see The phylogenetic relationships of Caenorhabditis and other rhabditids).
Most Caenorhabditis species form phoretic and/or necromenic associations with other soil invertebrates (see Ecology of Caenorhabditis species). In phoretic associations, hosts are used for transport between microenvironments. In necromenic associations, nematodes resume development and reproduce in the bacterial bloom that occurs following the host death. A variety of snails have been identified as hosts for C. briggsae dauer larvae (S. Baird, unpubl. obs.). C. briggsae dauers also have been obtained from soil and compost (Cutter et al., 2006). As dauers can persist for several months without feeding, generation times in wild populations are not known and may vary widely between and within populations.
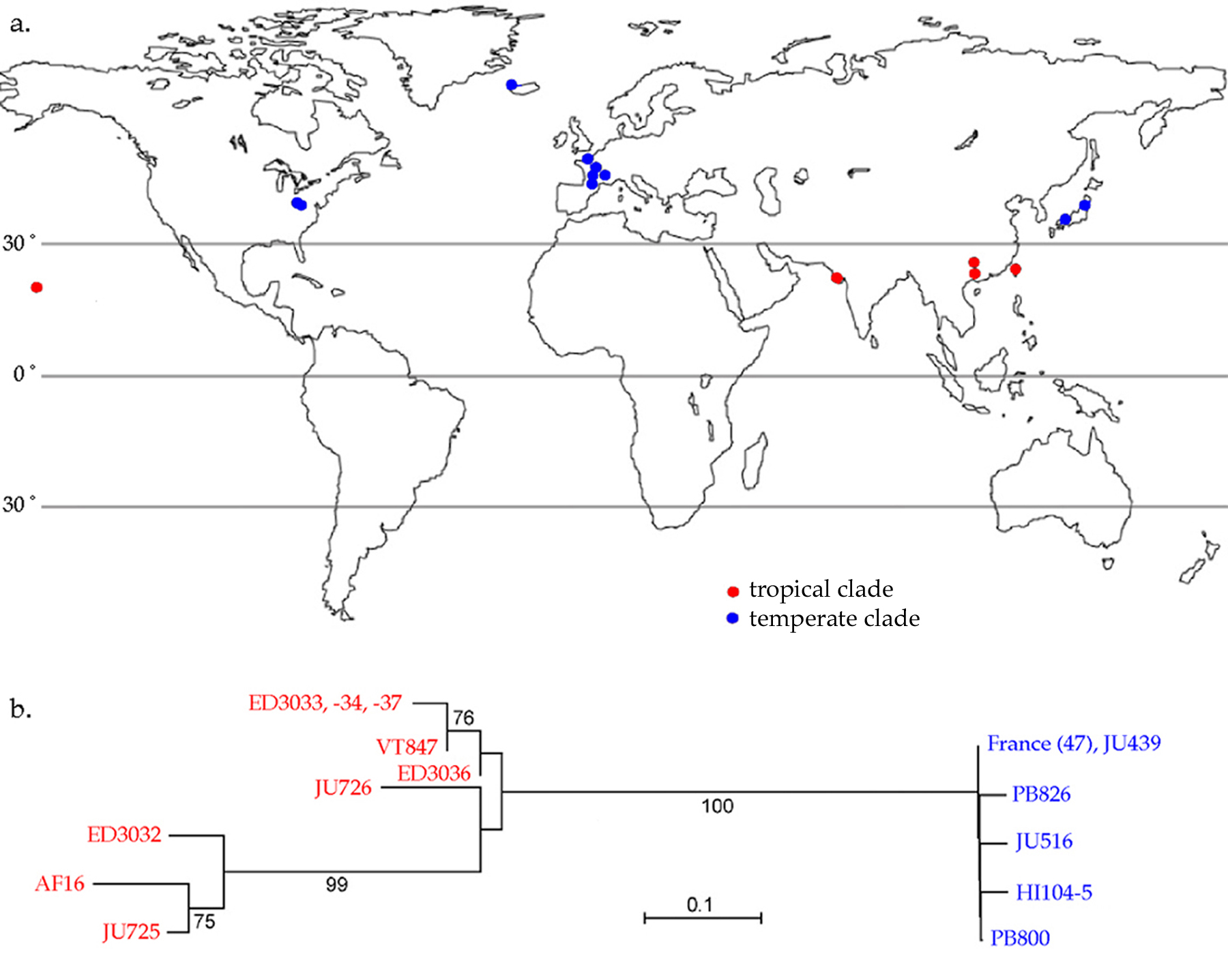
C. briggsae is a cosmopolitan species, having been obtained from collections in Asia, North America, and Europe (Figure 1A; Cutter et al., 2006). Phylogentic reconstructions of strains derived from these collections strongly support the division of C. briggsae into distinct tropical and temperate clades (Figure 1B; Graustein et al., 2002; Cutter et al., 2006; Baird and Hampton, unpubl. results). This clade structure should be considered when designing experiments, as tropical and temperate strains exhibit morphological and developmental differences. Consequently, different experimental results sometimes are obtained for tropical and temparate strains (Baird, 2001; 2002; Dellatre and Félix, 2001). Few collections have been made in the Southern hemisphere. However, C. briggsae strains have been obtained from Kenya and South Africa (E. Dolgin, personal communication).
The division of C. briggsae into tropical and temperate clades probably results from a recent expansion of the C. briggsae range into temperate latitudes. Nucleotide diversity within the temperate clade is very low and the coalescence time for this clade has been estimated to be 700 years (Cutter et al., 2006). This estimate was based on an effective population size (Ne) for the temaperate clade of 1,000 and a 60 day generation time (assuming that in wild populations, C. briggsae individuals spend most of their time as dauers).
|
Figure 1. Biogeography of Caenorhabditis briggsae. a) A map of tropical and temparate collection sites from which C. briggsae strains have been obtained. b) An unrooted reconstruction of C. briggsae phylogeny based on sequence comparisons at six loci. Adapted from Cutter et al. (2006).
Nomenclatural rules for C. briggsae, which currently are in flux, reflect the conflicting goals of providing unique durable gene names for mutationally identified genes while at the same time allowing for the identification of orthologs to C. elegans genes. The history of C. briggsae nomenclature rules and detailed nomenclatural suggestions can be found at the Caenorhabditis briggsae Research Page. One feature of this system is that C. briggsae gene names may change as orthology is determined. For example, the C. briggsae mip-1 gene was identified based on mutations that cause mutant animals to twitch uncontrollably. In C. elegans, this phenotype results commonly from mutations in unc-22. Hence, mip-1 may be the C. briggsae ortholog of unc-22. If this orthology is confirmed, mip-1 will be renamed Cb-unc-22. Confusion caused by changes in C. briggsae gene names is expected to be minimal as gene name histories will be archived in WormBase.
Alleles, strains, polymorphisms, rearrangements, transgenes, and other variants will be given unique identifiers following C. elegans nomenclatural conventions (Horvitz et al., 1979; CGC). These identifiers will include a lab strain or allele designation followed by a unique strain or allele number for that lab. For example, bd101 is a C. briggsae mutation that is present in several strains including PB101, PB107 and PB108. These names will be unique and durable.
Finally, it should be noted that a different convention is used by WormBase to name predicted C. briggsae genes. In this convention, each gene prediction gets a CBG (C. briggsae gene) prefix followed by a unique five digit identifier, e.g., CBG00123. Gene predictions in WormBase are continually refined as algorithms are improved. Therefore, predicted CBG gene names also may change over time.
The genome sequence of C. briggsae is that of the tropical strain, AF16 (Stein et al., 2003). To facilitate genetic studies, 13,000 randomly selected clones derived from a temperate strain, HK104, also have been sequenced. From these sequences over 26,000 single nucleotide polymorphisms (SNPs) have been identified giving a SNP density of greater than 0.2% (Hillier, Miller, Chinwalla, Fulton, Kolboldt, and Waterston, personal communication).
Two recombination maps have been constructed for C. briggsae. One is a SNP-based map that was constructed based on the segregation of SNP alleles in recombinant inbred lines (RIL) (Hillier, Miller, Chinwalla, Fulton, Kolboldt, and Waterston, personal communication). These RIL were derived from a cross between C. briggsae strains AF16 and HK104 (Baird et al., 2005). Each RIL was initiated with a single L4 F2 hermaphrodite and inbred through a single hermaphrodite per generation to F11. The current draft SNP map (v3.3) includes segregation data for 290 SNPs in 93 RIL. RIL genotypes for each SNP were determined using a fluorescent polarization-terminal dye incorporation (FP-TDI) assay (Hsu et al., 2001). This assay does not require any post-reaction product purification and has been adapted for the high-throughput genotyping of C. briggsae RIL. The other is a mutation-based map that was constructed based on the segregation of visible phenotypes in F2 progeny (B. Gupta, P. Sternberg, and D. Baillie, personal communication). Both maps identified six linkage groups. Integration of the SNP- and mutation-based maps is in progress (R. Miller and B. Gupta, personal communication). Nearly all C. briggsae induced mutations were derived from strain AF16. Therefore, map integration will be accomplished by mapping mutant alleles relative to HK104 SNPs. This will entail the genotyping of individual mutant F2s for multiple SNPs. To facilitate this, template DNA from F2 single-worm lysates will be amplified by whole genome amplification (Dean et al., 2001). Preliminary experiments have demonstrated the whole genome amplification provides template sufficient for several thousand genotyping reactions (S. Baird, unpubl results; B. Gupta and R. Miller, personal communication).
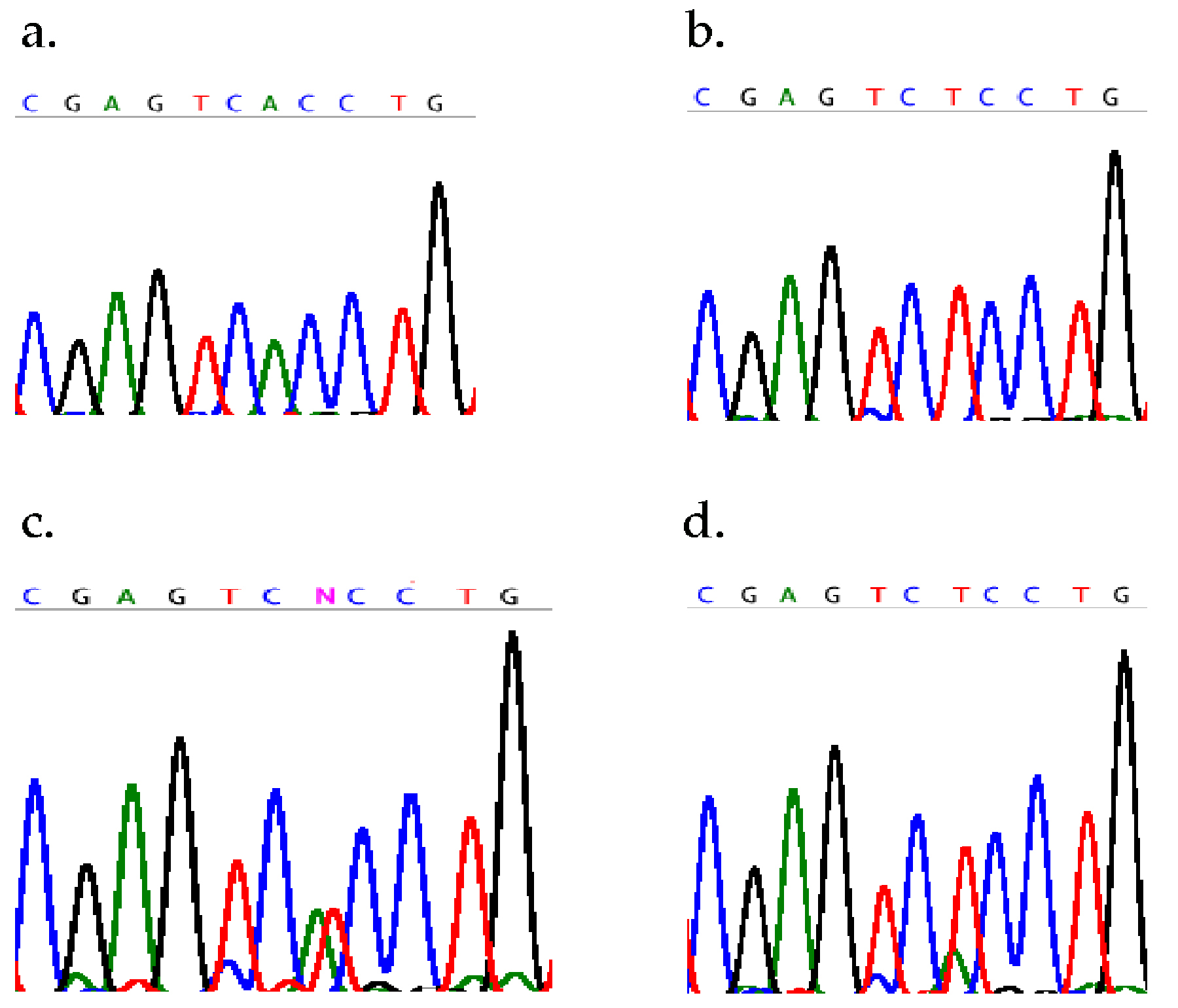
In C. briggsae, as in other organisms, most SNPs do not affect restriction endonuclease recognition sites. Therefore it is difficult and/or expensive to distinguish between AF16 and HK104 SNP alleles with using commonly available lab equipment. One way around this, at least for linkage determination, is bulk segregant analysis. In C. briggsae a bulk segregant assay is initiated by crossing an AF16-derived mutant strain to HK104. Pools of 10–25 mutant F2 progeny then are collected and lysed following a scaled-up (25 μl) single-worm protocol. Primers for these reactions are designed to amplify short (200–500 bp) products that contain a polymorphic nucleotide at least 50 nucleotides from either end. Amplification products are sequenced and polymorphisms are detected as ambiguities in the sequence traces. For unlinked loci, both alleles, i.e. an ambiguity, will be apparent in the sequence trace. For linked loci, only the AF16 allele will be present. In control reactions, a strong allelic bias was observed from lysates made from a 9:1 ratio of HK104 to AF16 hermaphrodites (Figure 2). In F2 progeny, a 9:1 ratio would be expected for loci located 10 mu from the selected mutation. Thus, two-fold redundant coverage of the C. briggsae genome can be achieved with as few as four SNPs per chromosome. This approach has been used successfully to identify loci involved in fitness loss in AF16::HK104 F2 hybrids (Hampton and Baird, unpubl. results).
|
Figure 2. Bulk segregant analysis of C. briggsae SNPs. Sequence traces from C. briggsae strains a) AF16 and b) HK104, showing an A/T polymorphisms. Sequence traces from lysates made from c) a mixture of five AF16 and five HK104 hermaphrodites and d) one AF16 hermaphrodite and nine HK104 hermaphrodites.
C. elegans and C. briggsae share a similar number of protein-coding genes (just under 20,000), with an estimate of 12,200 orthologous genes. Another 6,500 genes in C. briggsae have one or more C. elegans homolog, leaving about 800 genes as C. briggsae-specific (Stein et al., 2003). C. elegans–C. briggsae orthologs are annotated in Wormbase (http://www.wormbase.org/) according to the criteria of Stein et al. (2003). Each ortholog was identified either as the reciprocal top BLASTP match when queried against the genome of the other species, or was identified as a homolog (but not the reciprocal best match) with conserved synteny (gene order) with respect to other genes on the chromosome.
Although ortholog predictions by Stein et al. are fairly robust, false positives can result if best matches are not orthologous due to gene loss or divergence, or gene amplification events. Reciprocally, false negatives can result in the cases of rapid gene evolution or from errors in genome annotation. Consequently, researchers interested in a specific gene or set of genes will want to perform additional analysis to confirm orthology. Commonly, this can be achieved by phylogenetic reconstruction using all sequences of homologs available from different Caenorhabditis species, as well as an outgroup species. Homologs can be identified using BLAST to identify conserved domains as well as other genes (or translated gene products) that contain the domains. The relationship among the genes/proteins can be determined using a neighbor joining program with confidence assessed by bootstrapping methods (e.g., PHYLIP). True orthologs should exhibit fewer sequence differences and thus join, or cluster, with each other (e.g., Jovelin and Phillips, 2005; Nayak et al., 2005; Thomas, 2006).
Introduction of transgenes into C. briggsae allows for mutant rescue experiments, reporter transgene assays, and heterologous gene expression, both for ectopic expression studies, and inter-specific functional assays (e.g., Streit et al., 1999; Wang and Chamberlin, 2002; Wang and Chamberlin, 2004). High copy number extrachromosomal transgenes can be introduced into C. briggsae by microinjection of DNA into the hermaphrodite germline (see Transformation and microinjection) following protocols developed for C. elegans (Mello et al., 1991). Transformation markers from C. elegans that are effective in C. briggsae include ce-rol-6(d), ce-myo-2::gfp, ce-myo-3::gfp, ce-hsp-16-48::gfp, and ce-sur-5::gfp (Streit et al., 1999; Wang and Chamberlin, 2002) M.-A. Felix, personal communication). Extrachromosomal arrays can be integrated (see Transformation and microinjection) into the chromosome using 3000–4000 rad γ-irradiation and following C. elegans protocols (Inoue et al., 2002).
C. briggsae exhibits differences from C. elegans with respect to the efficacy of transgene formation, and they exhibit greater transgene silencing and mosaicism. In general, similar DNA mixtures and injection conditions result in 3-10 fold fewer stable transgenes in C. briggsae than in C. elegans, with reduced efficiency at both F1 and F2 generations (M.-A. Felix, personal communication). Although some of these differences may reflect the use of heterologous [usually C. elegans (Li et al., 2006)] DNA in transformation experiments, these results suggest that producing transgenes in C. briggsae will benefit from further optimization of protocols.
As in C. elegans, double stranded RNA-mediated gene interference (RNAi; see Reverse genetics) can provide a relatively easy method to assess the function of C. briggsae genes in vivo (e.g., Haag et al., 2002; Rudel and Kimble, 2001; Stothard et al., 2002; Streit et al., 1999). For this method, introduction of double-stranded RNA corresponding to a gene of interest results in the specific degradation of the corresponding mRNA, allowing phenotypic analysis of animals depleted for the gene. However, in contrast to C. elegans, an effective RNAi response is generally obtained in C. briggsae only when double stranded RNA is introduced directly into animals, such as by microinjection. C. briggsae is resistant to RNAi introduced by soaking or bacterial feeding methods (M. K. Montgomery, personal communication; C. Hunter, personal communication). This resistance reflects a difference between C. briggsae and C. elegans animals in availability of RNAi uptake and spreading mechanisms that allow RNAi transport into the animal and across epithelial boundaries. Consequently, RNAi can frequently be less effective in C. briggsae than C. elegans, and negative results or differences in RNAi phenotypes between the species must be interpreted with caution. Recent work has shown that the sid-2 gene required for environmental uptake of RNA by C. elegans is altered and not functional in C. briggsae. Consequently, introduction of ce-sid-2 as part of a transgene enhances RNAi uptake in C. briggsae, and confers sensitivity to RNAi by feeding onto this species (W. M. Winston, M. Sutherlin, A. J. Wright, E. Feinberg, and C. Hunter, unpublished results). We anticipate that these transgenic animals or other genetically modified or mutant C. briggsae strains will allow for enhanced RNAi response and a greater range of methods to introduce RNAi.
Although functional studies using RNAi are efficient, the gold standard in genetic analysis is chromosomal mutations. Strategies developed to isolate deletion alleles (see Reverse genetics) in C. elegans genes have been applied effectively to C. briggsae (Edgley et al., 2002), although the frequency of allele recovery may be somewhat lower than in C. elegans. Hill et al. (2006) report a single deletion allele each of Cb-fem-2 and Cb-fem-3 recovered from a library of 1.1 million EMS mutagenized gametes.
The Drosophila transposon Mos1 has been shown to mobilize and cause heritable mutations in C. elegans, providing a method for rapid tagging and recovery of mutant genes (Williams et al., 2005). Following a modified protocol (below), M.-A. Felix and her group have shown that Mos1 mutagenesis can be applied to C. briggsae as well. Although the SNP-based polymorphism map (v3.3) combined with genomic sequence will provide the tools for positional cloning of genes in C. briggsae, transposon tagging may allow a more efficient strategy for gene cloning in some cases.
In C. elegans, Mos1 mutagenesis is achieved using two transgenes: an extrachromosomal array that expresses the transposase under control of a heat-shock promoter, and a second transgene that includes the transposon (Bessereau et al., 2001). Expression of the transposase is induced in parental (P0) hermaphrodites, and then offspring are screened in the F1 or F2 for mutant phenotypes. Following outcross of the mutant allele, the tagged gene can be isolated using PCR. A detailed protocol for Mos1 mutagenesis is available (Bessereau et al., 2006).
In C. briggsae, Mos1 mutagenesis can be achieved following the C. elegans protocol, substituting transgenes mfEx18 (transposase) and mfEx24 (transposon) (M.A. Felix, personal communication). Heat-shock conditions are also modified to allow for the greater heat-tolerance of C. briggsae animals. Mos1 germline insertions and mutant alleles have been obtained using the following heat-shock treatment of P0 young adults:
37° C for one hour, 20° C for one hour, 37° for one hour, and 15° overnight.
37° C for 2.5 hours, 20° C for 35 minutes, 37° for one hour, and 25° overnight.
As the development of C. briggsae as a model system is in its infancy, information on its genetics and genomics is changing rapidly. The most valuable resources for staying up to date are a set of C. briggsae web sites that are the repository of considerable published and unpublished data.
This is the best site for C. briggsae experimental genetics. Of primary importance are archival lists of C. briggsae mutations, linkage data, and recombination maps based on visible mutations. This site also has links to a variety of other resources including a list of C. briggsae labs.
In addition to the SNP map, this site is a download site for C. briggsae sequence data, including strain-specific nucleotide polymorphisms.
C. briggsae genomics data have been incorporated into WormBase. Features include C. briggsae gene preditions, the identification of C. briggsae-C. elegans orthologs and syntenic alignments of C. briggsae and C. elegans.
The C. briggsae genome server offers sequence data downloads and a link to C. briggsae-specific BLAST server.
The CGC is a repository of C. briggsae wild isolates and mutant strains.
Baird, S.E. (2001). Strain-specific variation in the pattern of caudal papillae in Caenorhabditis briggsae (Nematoda: Rhabditidae); implications for species identification. Nematology 3, 373–376. Article
Baird, S.E. (2002). Haldane's Rule by sexual transformation in Caenorhabditis. Genetics 161, 1349–1353. Abstract Article
Baird, S.E., Davidson, C.R. and Bohrer, J.C. (2005). The genetics of ray pattern variation in Caenorhabditis briggsae. BMC Evol. Biol. 5, 3. Abstract Article
Bessereau, J.L., Wright, A., Williams, D.C., Schuske, K., Davis, M. W., and Jorgensen, E.M. (2001). Mobilization of a Drosophila transposon in the Caenorhabditis elegans germ line. Nature 413, 70–04. Abstract Article
Bessereau, J.L. (2006). Insertional mutagenesis in C. elegans using the Drosophila transposon Mos1: a method for the rapid identification of mutated genes. Methods Mol. Biol. 351, 59–73. Abstract
Cho, S., Jin, S.-W., Cohen, A. and Ellis, R.E. (2004). A phylogeny of Caenorhabditis reveals frequent loss of introns during nematode evolution. Genome Res. 14, 1207–1220. Abstract Article
Cutter, A.D., Félix, M.-A., Barriere, A. and Charlesworth, D. (2006). Patterns of nucleotide polymorphism distinguish temperate and tropical wild isolates of Caenorhabditis briggsae. Genetics 174, 2021–2031. Article
Dean, F.B., Nelson, J.R., Giesler, T.L., and Lasken, R.S. (2001). Rapid Amplification of plasmid and phage DNA using Phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 11, 1095–1099. Abstract Article
Dellatre, M. and Félix, M.-A. (2001). Polymorphism and evolution of vulval precursor cell lineages within two nematode genera, Caenorhabditis and Oscheius. Curr. Biol. 11, 631–643. Abstract Article
Edgley, M., D'Souza, A., Moulder, G., McKay, S., Shen, B., Gilchrist, E., Moerman, D., and Barstead, R. (2002). Improved detection of small deletions in complex pools of DNA. Nucleic Acids Res. 30, e52. Abstract Article
Graustein, A., Gasper, J.M., Walters, J.R. and Palopoli, M.F. (2002). Levels of nucleotide polymorphism vary with mating system in the nematode genus Caenorhabditis. Genetics 161, 99–107. Abstract Article
Haag, E.S., Wang, S., and Kimble, J. (2002). Rapid coevolution of the nematode sex-determining genes fem-3 and tra-2. Curr. Biol. 12, 2035–2041. Abstract Article
Hill, R.C., de Carvalho, C.E., Salogiannis, J., Schlager, B., Pilgrim, D., and Haag, E.S. (2006). Genetic flexibility in the convergent evolution of hermaphroditism in Caenorhabditis nematodes. Dev. Cell 10, 531–538. Abstract Article
Horvitz, H.R., Brenner, S., Hodgkin, J. and Herman, R.K. (1979). A uniform genetic nomenclature for the nematode Caenorhabditis elegans. Mol. Gen. Genet. 175, 129–133. Abstract Article
Hsu, T.M., Chen, X., Duan, S., Miller, R.D. and Kwok, P.-Y. (2001). Universal SNP genotyping assay fluorescence polarization detection. BioTechniques 31, 560–570. Abstract
Inoue, T., Sherwood, D.R., Aspock, G., Butler, J.A., Gupta, B.P., Kirouac, M., Wang, M., Lee, P.Y., Kramer, J.M., Hope, I., Burglin, T.R., and Sternberg, P.W. (2002). Gene expression markers for Caenorhabditis elegans vulval cells. Gene Expr. Patterns 2, 235–241. Abstract Article
Jovelin, R., and Phillips, P.C. (2005). Functional constraint and divergence in the G protein family in Caenorhabditis elegans and Caenorhabditis briggsae. Mol. Genet. Genomics 273, 299–310. Abstract Article
Kiontke, K., Gavin, N.P., Raynes, Y., Roehrig, C., Piano, F. and Fitch, D.H.A. (2004). Caenorhabditis phylogeny predicts convergence of hermaphroditism and extensive intron loss. Proc. Natl. Acad. Sci. U.S.A. 101, 9003–9008. Abstract Article
Li, X., Massey, H.C., Jr, Nolan, T.J., Schad, G.A., Kraus, K., Sundaram, M., and Lok, J.B. (2006). Successful transgenesis of the parasitic nematode Strongyloides stercoralis requires endogenous non-coding control elements. Int. J. Parasitol. 36, 671–679. Abstract Article
Mello, C.C., Kramer, J.M., Stinchcomb, D., and Ambros, V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970. Abstract Article
Nayak, S., Goree, J., and Schedl, T. (2005). fog-2 and the evolution of self-fertile hermaphroditism in Caenorhabditis. PLOS Biol. 3, e6. Abstract Article
Nigon, V. and Dougherty, E.C. (1949). Reproductive patterns and attempts at reciprocal crossing of Rhabditis elegans Maupas, 1900 and Rhabditis briggsae. J. Exp. Zool. 112, 485–503. Abstract Article
Rudel, D., and Kimble, J. (2001). Conservation of glp-1 regulation and function in nematodes. Genetics 157, 639–54. Abstract Article
Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., Coulson, A., D'Eustachio, P., Fitch, D.H., Fulton, L.A., Fulton, R.E., Griffiths-Jones, S., Harris, T.W., Hillier, L. W., Kamath, R.S., Kuwabara, P.E., Mardis, E.R., Marra, M.A., Miner, T.L., Minx, P., Mullikin, J.C., Plumb, R.W., Rogers, J., Schein, J.E., Sohrmann, M., Spieth, J., Stajich, J.E., Wei, C., Willey, D., Wilson, R.K., Durbin, R., and Waterston, R. (2003). The Genome Sequence of Caenorhabditis briggsae: A Platform for Comparative Genomics. PLOS Biol. 1. Abstract Article
Stothard, P., Hansen, D., and Pilgrim, D. (2002). Evolution of the PP2C Family in Caenorhabditis: Rapid Divergence of the Sex-Determining Protein FEM-2. J. Mol. Evol. 54, 267–282. Abstract Article
Streit, A., Li, W., Robertson, B., Schein, J., Kamal, I.H., Marra, M., and Wood, W.B. (1999). Homologs of the Caenorhabditis elegans masculinizing gene her-1 in C. briggsae and the filarial parasite Brugia malayi. Genetics 152, 1573–0084. Abstract Article
Thomas, J.H. (2006). Concerted evolution of two novel protein families in Caenorhabditis species. Genetics 172, 2269–2281. Abstract Article
Wang, X., and Chamberlin, H.M. (2002). Multiple regulatory changes contribute to the evolution of the Caenorhabditis lin-48 ovo gene. Genes Dev. 16, 2345–2349. Abstract Article
*Edited by Ralf J. Sommer. WormMethods editor Victor Ambros. Last revised November 6, 2006. Published December 18, 2006. This chapter should be cited as: Baird, S.E. and Chamberlin, H.M. Caenorhabditis briggsae methods (December 18, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.128.1, http://www.wormbook.org.
Copyright: © 2006 Scott E. Baird and Helen M. Chamberlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: scott.baird@wright.edu or chamberlin.27@osu.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.