Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Cell isolation and culture are essential tools for the study of cell function. Isolated cells grown under controlled conditions can be manipulated and imaged at a level of resolution that is not possible in whole animals or even tissue explants. Recent advances have allowed for large-scale isolation and culture of primary C. elegans cells from both embryos and all four larval stages. Isolated cells can be used for single-cell profiling, electrophysiology, and high-resolution microscopy to assay cell autonomous development and behavior. This chapter describes protocols for the isolation and culture of C. elegans embryonic and larval stage cells. Our protocols describe isolation of embryonic and L1 stage cells from nematodes grown on high-density NA22 bacterial plates and isolation of L2 through L4 stage cells from nematodes grown in axenic liquid culture. Both embryonic and larval cells can be isolated from nematode populations within 3 hours and can be cultured for several days. A primer on sterile cell culture techniques is given in the appendices.
The methodology of culturing isolated cells has been available for over a century. The first reported primary cultures were explants of frog neuronal fibers (Harrison et al., 1907) and dissociated sponges (Wilson, 1907) that grew and differentiated over the course of several days. By the 1950s, the introduction of protease digestion, defined media, and antibiotics made vertebrate cell culture a more widely accessible technique.
Aside from amoeba-like nematode sperm (Klass and Hirsh, 1981), C. elegans cells have proved to be far more difficult to isolate and culture than vertebrate or insect cells. The main difficulty has been that the chitinous shell of embryos and the tough outer cuticle of larval and adult worms are resistant to cell and tissue explantation. Early methods involving mechanical segregation of embryonic cells by cutting and extruding eggs (Laufer et al., 1980), or laser cutting followed by extrusion (Laufer, 1982), or shearing eggs by passage through a Hamilton syringe (Cowan and McIntosh, 1985) provided enough viable blastomeres to follow individual embryos and to test culture media. However, yields were low and few cells survived the process. Chemically dissolving the egg shell with a mixture of chitinase and chymotrypsin (Edgar and McGhee, 1988) followed by gently pulling the embryos through a glass needle to disrupt the vitelline layer (Goldstein, 1992) greatly increased the viability of embryonic cells. Subsequent work found that more vigorous pipetting after egg shell digestion (Bloom, 1993: http://hdl.handle.net/1721.1/12667; Christensen and Strange, 2001; Christensen et al., 2002; Strange et al., 2007) could extract large populations of embryonic cells for culture.
In contrast to embryos, the tough cuticle of larval and adults nematodes formed an almost impenetrable barrier to post-embryonic cell culture. Although releasing eggs from gravid adults with hypochlorite and a strong base is a mainstay technique of embryo isolation (Khan and Mcfadden, 1980: http://dx.doi.org/10.1163/187529280X00189), adult and larval cells are dissolved in the process. Researchers can gain access to adult cells for electrophysiology by immobilizing and filleting individual nematodes (Richmond and Jorgensen, 1999), but the technique is challenging and individual cells cannot be separated and cultured. The introduction of gentle chemical disruption of the larval cuticle and subsequent release of cells through repetitive pipetting (Zhang et al., 2011) has now allowed primary culture of C. elegans cells from all four larval stages.
The protocols collected here provide comprehensive techniques and media recipes for large-scale isolation and culture of primary embryonic and larval cells from C. elegans. We suggest equipment and supplies for cell culture in Appendix A and Appendix B. We include common cell culture techniques in Appendix C for those unfamiliar with working in a sterile culture hood.
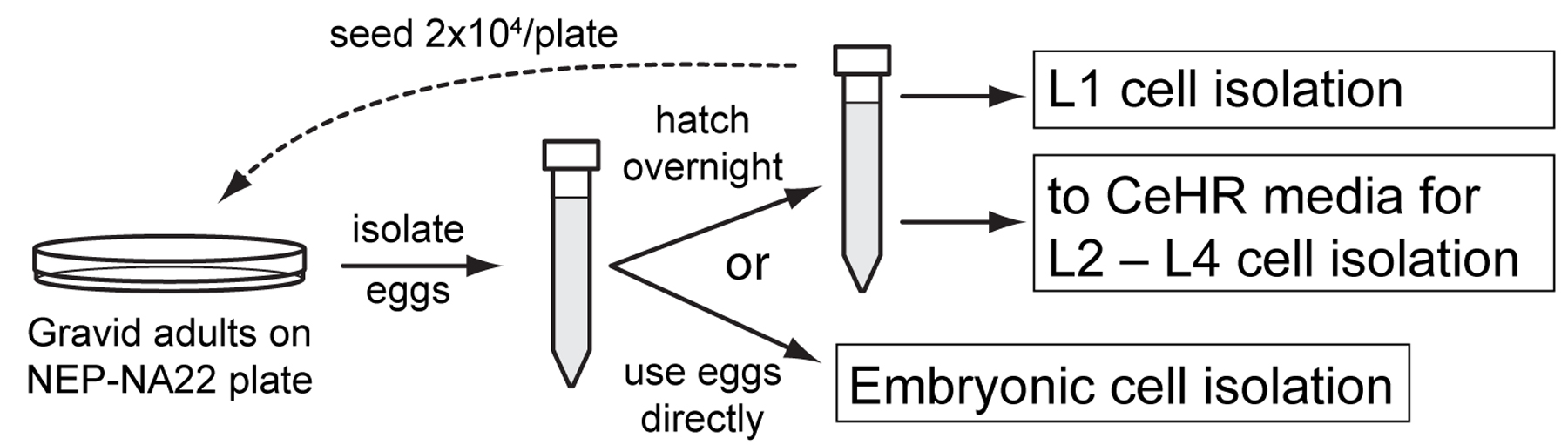
Both embryonic and larval cell isolations require 30 to 50 µl of compacted eggs or larvae per preparation. NA22 bacteria grow in thick layers on peptone-augmented agar plates and provide a richer food source than do OP50 bacteria, which is standardly used for nematode maintenance. A large number of worms can obtained by growing synchronized worms at high densities on 100 mm diameter NEP plates seeded with NA22 bacteria (NEP-NA22). Gravid adults from these plates are bleached in an alkaline solution (egg preparation) to obtain sterile eggs that can be used directly for embryonic cell isolation. Alternatively, eggs may be hatched overnight to produce synchronized L1 larvae for cell isolation or for synchronized axenic culture to obtain L2 through L4 stage cells.
Sterile petri dishes, 100 mm diameter
NaCl
Peptone
Agar
Cholesterol, 5 mg/ml (w/v) in ethanol. Do not autoclave.
1 M magnesium sulfate (MgSO4) in ddH2O, autoclaved.
1 M potassium phosphate pH 6.0. Mix 35.6 g K2HPO4 with 108.3 g KH2PO4 in 0.8 L ddH2O, and adjust pH to 6.0. Bring to 1 L in ddH2O and autoclave.
Nystatin Suspension (Sigma N1638). Fungicide, 10,000 units/ml in DPBS.
LB Broth – Miller formulation. NA22 bacteria grow well in moderate or high salt formulations of lysogeny broth (10g/L NaCl). 2X YT media may also be used to grow NA22 bacteria.
NA22 bacteria. NA22 bacteria may be obtained from the Caenorhabditis Genetics Center (CGC, http://www.cbs.umn.edu/cgc).
Dissolve the following components in ddH2O and bring to 1 L with ddH2O
| Component | Qty/1 L |
|---|---|
| NaCl | 1.2 g |
| peptone | 20 g |
| agar | 25 g |
Add a large stir bar and autoclave on a liquid cycle for 30 to 40 minutes.
Cool to approximately 60 °C while stirring.
Using aseptic techniques, add the following stock solutions
| Stock solution | Qty/1 L |
|---|---|
| 5 mg/ml cholesterol (in ethanol) | 1 ml |
| 1 M MgSO4 (autoclaved) | 1 ml |
| 1 M potassium phosphate pH 6.0 (autoclaved) | 25 ml |
| 100x Nystatin (stock 10,000 U/ml, final 100 U/ml) | 10 ml |
Pour 18 ml of medium each into 100 mm diameter petri dishes.
Aseptically inoculate 30 to 50 ml of LB broth with a colony of NA22 bacteria and grow overnight at 37 °C.
To each NEP plate, add 1 ml of NA22 culture and spread over the entire NEP plate surface.
Let seeded plates stand upright at room temperature for 3 to 4 days or until dry.
Invert seeded plates and store at 4 °C.
NaCl
Potassium phosphate monobasic (KH2PO4)
Sodium phosphate dibasic (Na2HPO4)
M9 media (recipe below)
1 M magnesium sulfate (MgSO4) in ddH2O, autoclaved.
Dissolve the following components in ddH2O and bring to 1 L with ddH2O
| Component | Qty/1 L |
|---|---|
| Potassium phosphate monobasic (KH2PO4) | 3 g |
| NaCl | 5 g |
| Sodium phosphate dibasic (Na2HPO4) | 6 g |
Autoclave the solution for 30 minutes on a liquid cycle.
Allow solution to cool to room temperature and then add
| Stock solution | Volume |
|---|---|
| 1 M Magnesium sulfate (MgSO4) | 1 ml |
Note: MgSO4 will precipitate out of solution at temperatures above 55 °C. Do not add MgSO4 before autoclaving M9 or before the autoclaved liquid has cooled.
Store at room temperature
Seed NEP-NA22 plates by chunking worms from NGM-OP50 plates or any other source.
Grow worms for several generations until plates are full of gravid adults.
Wash worms from plate by pipetting 10 ml of ddH2O directly onto the plate surface. Repeat and collect suspended worms in a 15 ml conical tube. Worms may be collected from several plates by washing the next plate with the same suspension of worms.
Settle the worms for approximately 2 minutes at room temperature in a 15 ml conical tube.
Remove excess ddH2O, leaving 7 ml of worm suspension.
Follow protocol 2.3 (Preparing eggs or hatched larvae) to produce synchronized L1 in M9 media.
The next day, count the hatched worms and plate 20,000 of synchronized L1 per plate onto NEP-NA22 plates.
Grow at 20 to 25 °C until the worms reach gravid adult stage.
Note: Growing synchronized worms at 20,000 per plate ensures that worms reach adult stage just before the plate starts to starve. One plate of such culture typically yields 100,000 to 200,000 eggs, which can be used to seed 5 to 10 NEP-NA22 plates. If an insufficient number of worms are obtained in the first round, repeat steps 3 through 8 above.
5 M sodium hydroxide (NaOH) in ddH2O.
Fresh sodium hypochlorite (household bleach). Purchase a fresh bottle of bleach every two weeks.
Start with gravid adults suspended in 7 ml of ddH2O.
To lyse worms and obtain eggs, add 1 ml of 5 M NaOH and 2 ml of fresh sodium hypochlorite (bleach) to the worm suspension. Vortex the suspension continuously at the highest speed of a laboratory vortex mixer for the entire 5 minute incubation. Do not exceed 5 minutes of lysis.
Note: Do not contaminate the outside rim of the conical tube or the cap with bacteria in the worm suspension. These bacteria do not contact the bleach/NaOH solution and may contaminate subsequent cultured cells.
Pellet the lysed worms at 1,300 g for 1 minute in a clinical centrifuge and discard the supernatant.
Note: Some egg preparation protocols subsequently separate eggs from lysed adult carcasses by floating the eggs on a layer of sucrose. We have found that the sucrose flotation step can be skipped with no effect in cell isolation outcome as long as adults are vortexed continuously during the alkaline hypochlorite treatment. Continuous vortexing sufficiently breaks down adult carcasses with little harm to the eggs.
Wash the pellet 3 times by resuspending in 10 ml sterile ddH2O and centrifuging at 1,300 g for 1 minute each. Insure that the pellet is thoroughly distributed before centrifugation.
Note: Eggs from this step can be used directly for embryonic (5.1) or L1 (6.2) cell isolation if the washing steps are done under sterile conditions in a cell culture hood.
To synchronize worms by hatching, resuspend eggs with 10 ml of M9 in a 15 ml conical tube and rotate at room temperature overnight.
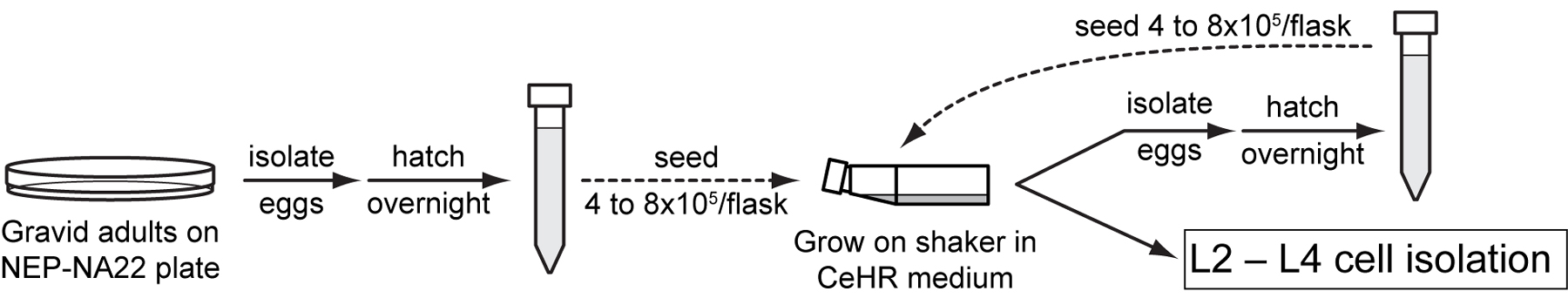
C. elegans larvae grown on bacteria cannot be used directly for larval cell isolation. Because the limited antibiotics used in cell culture media are bacteriostatic rather than bactericidal, contamination is always a risk. To overcome this problem CeHR medium, a bacteria-free worm culture medium developed by Eric Clegg's group (personal communication, http://elegans.som.vcu.edu/ECWM/2002/abstracts.pdf) is used for growing larvae for cell isolation. The CeHR basal medium, consisting of growth factors, nucleic acids and minerals, can be prepared separately and stored for an extended time period as frozen aliquots. Complete CeHR medium is prepared by adding skim milk to thawed CeHR basal medium. Switching from a bacterial diet to axenic medium is simply accomplished by inoculating CeHR medium with L1 larvae from a sterile egg preparation of worms grown on bacteria. C. elegans develop in CeHR axenic medium at a pace similar to growth on NA22 or OP50 bacterial plates. The recipe and assembly steps for CeHR medium shown below are based on a protocol from Nass and Hamza (Nass and Hamza, 2007). Although the recipe is laborious, CeHR medium for approximately 50 flasks of nematodes can be prepared in one day and stored in frozen aliquots.
1 M KOH
0.1 M NaOH
Concentrated HCl
Ethanol
High temperature ultra-pasteurized (UHT) skim milk. UHT sterilized milk has a long shelf life. It is typically sold in aseptic cartons in most grocery stores throughout the world. In the U.S.A., Horizon brand boxed milk is available in most grocery stores.
Additional components (listed below). Additional materials along with suggested sources and catalog numbers are listed in the tables below. “Storage” refers to the temperature at which the dry chemical is stored according to the manufacturer's recommendations. RT refers to room temperature.
Note: CeHR components are mixed under non-sterile conditions unless otherwise indicated. The final medium is assembled and filtered under sterile conditions.
To 60 ml ddH2O add the following components.
| Component | g | Storage | Source | Cat. # |
|---|---|---|---|---|
| N-Acetylglucosamine | 0.150 | 4 °C | Calbiochem | 1079 |
| L-Alanine | 0.150 | RT | Calbiochem | 1250 |
| Nicotinamide | 0.075 | RT | Sigma | N-3376 |
| D-Pantethine | 0.0375 | 4 °C | Sigma | P-2125 |
| D-Pantothenate (Ca) | 0.075 | 4 °C | Sigma | P-5155 |
| Pteroylglutamic Acid (Folic Acid) | 0.075 | RT | ACROS | 21663-0100 |
| Pyridoxamine.2HCl | 0.0375 | −20 °C | Sigma | P-9158 |
| Pyridoxine.HCl | 0.075 | RT | Sigma | P-6280 |
| Riboflavin 5-PO4(Na) | 0.075 | −20 °C | Sigma | R-7774 |
| Thiamine.HCl | 0.075 | RT | Sigma | T-1270 |
To 5 ml of 1 M KOH add the following components.
| Component | g | Storage | Source | Cat. # |
|---|---|---|---|---|
| p-Aminobenzoic Acid | 0.075 | 4 °C | Sigma | A-9878 |
| Biotin | 0.0375 | 4 °C | Sigma | B-4639 |
| Cyanocobalamine (B-12) | 0.0375 | 4 °C | Sigma | V-2876 |
| Folinate (Ca) | 0.0375 | RT | Sigma | F-7878 |
| Nicotinic acid | 0.075 | RT | Sigma | N-0761 |
| Pyridoxal 5′-phosphate | 0.0375 | −20 °C | Sigma | P-3657 |
To 1 ml of ethanol add the following component.
| Component | g | Storage | Source | Cat. # |
|---|---|---|---|---|
| (±) α-L-lipoic acid, oxidized form | 0.0375 | 4 °C | Sigma | T-1395 |
Mix solutions from steps 1 through 3, and bring to 100 ml with ddH2O.
Freeze as 10 ml aliquots in 15 ml conical tubes and store at −20 °C.
To 60 ml ddH2O add the following components in order.
| Component | g | Storage | Source | Cat. # |
|---|---|---|---|---|
| Adenosine 5′-PO4 (Na) | 1.74 | −20 °C | Sigma | A-1752 |
| Cytidine 5′-PO4 (Na) | 1.84 | 4 °C | Sigma | C-1006 |
| Guanosine 2′- & 3′-PO4 (Na) | 1.82 | −20 °C | Sigma | G-8002 |
| Uridine 5′-PO4 (Na) | 1.84 | −20 °C | Sigma | U-6375 |
| Thymine (add last) | 0.63 | RT | Sigma | T-0376 |
Bring to 100 ml with ddH2O.
Freeze in 10 ml aliquots in 15 ml conical tubes and store at −20 °C.
To 150 ml ddH2O add the following components.
| Component | g | Storage | Source | Cat. # |
|---|---|---|---|---|
| MgCl2.6H20 | 0.82 | RT | Sigma | M-2393 |
| Sodium Citrate | 0.58 | RT | Sigma | S-4641 |
| Potassium Citrate. H2O | 0.98 | RT | Sigma | P-1722 |
| CuCl2.2H2O | 0.014 | RT | Fisher | C455-500 |
| MnCl2.4H2O | 0.04 | RT | Fisher | M87-100 |
| ZnCl2 | 0.02 | RT | Sigma | Z-0152 |
| Fe(NH4)2(SO4)2.6H2O | 0.12 | RT | Sigma | F-1543 |
| CaCl2.2H2O | 0.04 | RT | Fisher | C70-500 |
Bring to 200 ml with ddH2O and store at 4 °C.
Add 19.5 mg of hemin chloride to 10 ml of 0.1 M NaOH.
| Component | g | Storage | Source | Cat. # |
|---|---|---|---|---|
| Hemin chloride | 0.0195 | RT | Frontier Scientific | H651-9 |
Adjust pH to 8.0 with concentrated HCl.
Bring to 15 ml with 0.1 M NaOH. The final concentration will be 2 mM (1.3 mg/ml).
Use immediately or store at −20 °C in dark or foil covered containers for up to 2 weeks.
Add 50 mg of cholesterol to 10 ml of ethanol for a final concentration of 5 mg/ml.
| Component | g | Storage | Source | Cat. # |
|---|---|---|---|---|
| Cholesterol | 0.05 | RT | J.T. Baker | F676-05 |
Purchase the following amino acid mixtures and store at 4 °C.
| Component | Storage | Source | Cat. # |
|---|---|---|---|
| MEM Essential Amino Acid Mix (50x) | 4 °C | GIBCO | 11130-051 |
| MEM Non-essential Amino Acid Mix (100x) | 4 °C | GIBCO | 11140-050 |
Note: Only open the bottles in the culture hood.
Bring 6.13 g of KH2PO4 to 100 ml in ddH2O for a final concentration of 0.45 M. Store at 4 °C.
Disinfect the unopened box by spraying the outside with 70% ethanol. Wipe with Kimwipes (Kimberly-Clark) or equivalent low lint wipe.
Move the box to the culture hood and open it.
Divide the milk into 10 ml aliquots in 15 ml conical tubes.
Store at −20 °C and thaw as needed.
Make each of the following components separately in the indicated final volume of ddH2O.
| Component | g | Final Volume | Stock | Source | Cat. # |
|---|---|---|---|---|---|
| Choline di-acid citrate | 0.885 | 15 ml | 4 °C | Sigma | C-2004 |
| i-Inositol | 0.648 | 15 ml | RT | Sigma | I-5125 |
| D-Glucose | 13.15 | 50 ml | RT | Sigma | G-7021 |
| HEPES (Na salt) | 3.9 | 15 ml | RT | Sigma | H-3784 |
| Lactalbumin hydrolysate | 4.25 | 25 ml | 4 °C | Sigma | L-9010 |
Note: Perform all procedure in the culture hood under sterile conditions.
Place a 500 ml or larger, 0.22 μm bottle-top vacuum filter in the culture hood.
Disconnect the vacuum or leave the vacuum off.
Add following solutions to the reservoir in order.
| Solution | Volume |
|---|---|
| Choline | 5 ml |
| Vitamin mix | 5 ml |
| i-Inositol | 5 ml |
| Hemin chloride | 5 ml |
| ddH2O | 125 ml |
Note: Hemin chloride will precipitate after filtration. The subsequent nucleic acid mix will re-solubilize this precipitate.
Connect or turn on the vacuum and wait for solutions to pass through the filter.
Disconnect or turn off the vacuum and add
| Solution | Volume |
|---|---|
| Nucleic acid mix | 10 ml |
Swirl the vacuum unit to solubilize any unfiltered hemin chloride precipitate.
Connect or turn on the vacuum and wait for solutions to pass through the filter. Disconnect or turn off the vacuum.
Add the following components to the reservoir in order.
| Solution | Volume |
|---|---|
| Mineral mix | 50 ml |
| Lactalbumin | 10 ml |
| MEM Essential Amino Acid Mix (50x) | 10 ml |
| MEM Non-essential Amino Acid Mix (100x) | 5 ml |
| 0.45M KH2PO4 | 10 ml |
| d-Glucose | 25 ml |
| HEPES (Na salt) | 5 ml |
| ddH2O | 125 ml |
| Cholesterol | 0.5 ml |
Connect or turn on the vacuum and wait for solutions to pass through the filter. Disconnect or turn off the vacuum.
Remove the filter unit, cap the bottle with a sterile top, and gently swirl the solution until mixed.
The above procedure yields approximately 395 ml CeHR basal medium. Aliquot 8 ml each to several 15 ml conical tubes. Store aliquots at −20 °C.
CeHR Media (protocol 3.1).
T25 tissue culture flasks (25 cm2 surface), uncoated, with vented caps.
Incubated orbital shaker.
Follow protocol 2.2 (Propagating and synchronizing worms on NEP-NA22 plates).
Follow protocol 2.3 (Preparing eggs or hatched larvae) to produce synchronized L1 in sterile M9 media.
Note: Eggs are initially sterilized during the alkaline hypochlorite digestion. All subsequent steps must be performed in a cell culture hood with sterile media.
Allow worms to hatch to L1 stage in sterile M9 media overnight on a laboratory rotator.
Optional: Prepare a new 15 ml conical tube by adding 0.5 ml of sterile ddH2O. Mark the level of the water, remove the water, and transfer 10 ml of newly hatched larvae to this tube.
Sterilely remove 5 µl of worm suspension. Count the number of worms under a dissecting microscope and divide by 0.005 to estimate the density of synchronized larvae per ml in the original suspension.
Pellet larvae at 1,300 g for 1 minute in a clinical centrifuge.
Remove most of the supernatant to leave 0.5 ml of total volume and resuspend the pellet. Multiply the density calculated from step 5 by 20 to estimate the density of concentrated larvae.
Thaw one aliquot of CeHR basal medium per flask. Add 2 ml of freshly thawed skim milk to every 8 ml of CeHR basal medium. Mix and transfer 10 ml of complete CeHR medium into a T25 tissue culture flask.
Seed worms at 40,000 to 80,000 larvae per flask of CeHR medium.
Grow worms at 70 RPM in an incubated orbital shaker at 22 °C until gravid adults begin to lay eggs.
Note: Sterile CeHR medium (with milk) is cloudy but will clear as worms consume the nutrients. Check the flask frequently after seeding. If the media becomes cloudier over time, it is likely contaminated. Check a small aliquot for contamination and discard the flask according to your institution's safety policies.
Once gravid adults form, sterilely collect the CeHR suspension in a 15 ml centrifuge tube.
Note: If the incubated shaker is also used to culture bacteria, thoroughly disinfect the outside of the flask with 70% ethanol before transferring to the cell culture hood.
Centrifuge worms at 1,300 g for 1 minute, remove the supernatant, and resuspend with 7 ml of sterile ddH2O.
Follow protocol 2.3 (Preparing eggs or hatched larvae) to produce synchronized L1 in sterile M9 media.
Note: Worms harvested from CeHR medium can be frozen using standard procedures.
Repeat steps 3 through 8 and inoculate new CeHR culture flasks by seeding worms 40,000 to 80,000 larvae per flask of CeHR medium.
Grow larvae on a shaker to the desired stage for larval cell preparation (Figure 3). Alternatively, allow worms to grow to gravid adult stage and prepare and hatch eggs in M9 media. Seed new flasks of CeHR media with synchronized larvae to maintain a sterile, synchronized population of nematodes for subsequent experiments.
Note: The first generation of larvae will grow slower in CeHR (7 to 10 days) and unsynchronized as worms adapt to the new environment. The second generation grows faster and synchronized, reaching gravid adult stage in approximately 3 to 4 days (Nass and Hamza, 2007). Do not use the first generation of larvae previously grown on NEP plates for cell isolation. Use the second generation of worms adapted to CeHR for larval cell isolation.
|
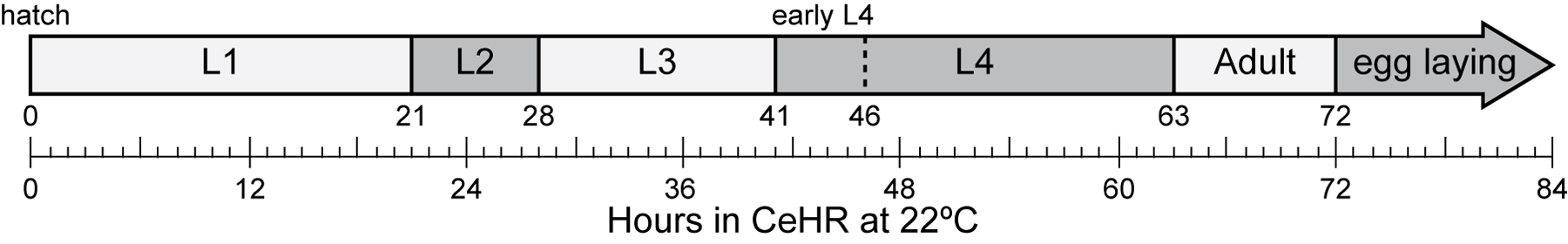
Figure 3. Approximate growth rate of nematodes at 22 °C after adaptation to CeHR (Szilagyi et al., 2006: http://www.dtic.mil/cgi-bin/GetTRDoc?AD=ADA469210).
A number of media formulations have been used to support the survival of isolated C. elegans cells. Like insect culture media (Schlaeger, 1996), nematode cell culture media are maintained at higher osmolarities than those used for vertebrate cell culture. Although intact nematodes can survive osmotic stresses (Lamitina et al., 2004; Choe and Strange, 2008), cultured C. elegans cells lack the secretory cells (renal system) of the whole animal and thus may be sensitive to osmotic stress. An osmotic pressure of 340 mOsm, approximately twice what is used for vertebrate cell culture (Schlaeger, 1996), is ideal for supporting the survival and differentiation of C. elegans embryonic cells (Christensen et al., 2002).
The first reported C. elegans cell culture medium was developed by Lois Edgar (Edgar's Growth Media, EGM) for growing embryonic blastomeres (Edgar and McGhee, 1988; Edgar and Goldstein, 2012). EGM is a complex and rich medium containing amino acids, salts, and nucleic acid precursors, with fats and cholesterol provided by addition of egg yolk. EGM osmolarity is supplemented by addition of polyvinylpyrrolidone (PVP) and inulin. In an effort to simplify EGM, Bloom and Horvitz (Bloom, 1993: http://hdl.handle.net/1721.1/12667) tested several formulas based on commercially available insect media. They found that L-15 supplemented with nucleic acid precursors, fetal bovine serum to provide growth factors, fats, and cholesterol, and PVP and inulin to increase osmolarity, provided an optimal mix for supporting neuronal outgrowth. Christensen et al. (Christensen et al., 2002) further simplified the L-15-based embryonic growth media by dispensing with nucleic acid precursors and replacing PVP and inulin with sucrose to increase osmolarity. The cell culture medium that we describe below is the Christensen L-15-based medium, with osmolarity adjusted to the recommended 340 mOsm.
L-15 insect media (Invitrogen 21083-027).
Fetal Bovine Serum (FBS, Invitrogen 16000-077).
Penicillin-Streptomycin solution (Sigma P4458). 5,000 units penicillin and 5 mg streptomycin/ml
Sucrose, 60% w/v in ddH2O. Autoclave and use aseptically.
Osmometer calibration standards.
Equilibrate the osmometer to room temperature overnight.
Thaw FBS overnight at 4 °C.
Assemble the following components sterilely in the cell culture hood.
| Component and final concentration | Stock | Qty/500 ml |
|---|---|---|
| L-15 insect media | 1x | 450 ml |
| 10% FBS | 100% | 50 ml |
| 50 U/ml penicillin + 50 μg/ml streptomycin (Sigma P4458) | 100x | 5 ml |
Note: Minimize freezing and thawing of FBS. If using a new bottle of FBS, use the culture hood to sterilely pour any remaining FBS into 50 ml tubes (protocol 11.10 in Appendix C) and freeze the aliquots. Thereafter, thaw one aliquot at a time.
Remove the L15/FBS medium from the culture hood.
Calibrate the osmometer according to the manufacturer's instructions.
Using bench-top aseptic methods (flame, autoclaved pipette tips), adjust the osmolarity of the L15/FBS to 340±5 mOsm with 60% sucrose. Approximately 8.5 ml of sucrose will be required.
In a tissue culture hood, re-sterilize the medium with a 0.22 μm vacuum filter unit (protocol 11.7 in Appendix C).
Store at 4 °C.
1 M HEPES in ddH2O, pH 7.3. Store at 4 °C.
2 M NaCl in ddH2O. Store at room temperature.
2 M KCl in ddH2O. Store at room temperature.
1 M CaCl2 in ddH2O. Store at room temperature.
1 M MgCl2 in ddH2O. Store at room temperature.
Osmometer calibration standards.
Assemble the following components at room temperature and bring to 500 ml with ddH2O
| Components and final concentration | Stock | Qty/500 ml |
|---|---|---|
| 25 mM HEPES pH 7.3 | 1 M | 12.5 ml |
| 118 mM NaCl | 2 M | 29.5 ml |
| 48 mM KCl | 2 M | 12 ml |
| 2 mM CaCl2 | 1 M | 1 ml |
| 2 mM MgCl2 | 1 M | 1 ml |
Calibrate the osmometer according to the manufacturer's instructions.
Adjust osmolarity to 340±5 mOsm by diluting with sterile ddH2O.
In a tissue culture hood, sterilize the buffer with a 0.22 μm vacuum filter unit (protocol 11.7 in Appendix C).
Store at 4 °C.
Coverslips. Choose a size and thickness appropriate for the microscope.
Sterile petri dishes, 35 mm diameter.
1 M HCl. Concentrated HCl is 12.2 M. Slowly add 82 ml of concentrated HCL (drop-wise) to 0.8 L of ddH2O while stirring. Bring to 1 L with additional ddH2O. Store at room temperature in a glass bottle.
70% v/v ethanol in ddH2O. Make fresh each time.
Absolute ethanol.
Heated water bath
Water bath sonicator
Large Pyrex® or metal container for autoclaving coverslips.
Aluminum foil.
Fill a glass beaker or wide-mouth glass jar with 1 M HCl, separate coverslips, and drop them one at a time into the beaker.
Note: Clean one box (1 oz) of coverslips at a time at most.
Heat the beaker at 50 to 60 °C for 4 to 16 hrs.
Gently pour off the HCl and discard according to your institution's safety policies.
Rinse the coverslips once by swirling with ddH2O, allow the coverslips to settle and pour off water.
Fill the container with ddH2O and sonicate in a water bath for 30 minutes. Repeat sonication once with fresh ddH2O and discard the ddH2O.
Fill the container with freshly made 70% ethanol and sonicate for 30 minutes. Discard the ethanol.
Fill the container with absolute ethanol and sonicate for 30 minutes. Discard the ethanol.
Note: “Drying”, or removing the coverslips of residual water with absolute ethanol must be done stepwise. Do not switch coverslips directly from ddH2O to absolute ethanol.
Fill the container with absolute ethanol, cover tightly, and store for up to 1 year. Do not store coverslips and ethanol in plastic containers.
To sterilize coverslips for use, remove coverslips one at a time from the ethanol with forceps and layer them at the bottom of a large Pyrex beaker, glass petri dish, rectangular metal dish, or empty pipette tip box. Multiple layers may be separated with sheets of aluminum foil.
Cover the container with aluminum foil, add a strip of autoclave tape, and autoclave on a dry/gravity cycle
Note: Many coverslip sterilization protocols recommend flame-drying coverslips. Open flames are no longer recommended for use inside of a cell culture hood.
Open the autoclaved packet of coverslips in the cell culture hood and decontaminate forceps with 70% ethanol. Using the forceps, place coverslips in sterile 35 mm petri dishes. Store the dishes in the culture hood.
Peanut lectin (Arachis hypogaea agglutinin, Sigma L0881, 5 mg)
Acid washed coverslips in 35 mm petri dishes. Individual coverslips are ideal for subsequent fixation and antibody staining after several days in culture.
OR
Glass-bottom culture dishes (Mat Tek Corporation, P35G-1.0-14-C). These 35 mm diameter petri dishes have a coverslip affixed across a hole in the bottom of the dish. They can be used for high-resolution imaging of live nematode cells with an inverted microscope. Cells in glass-bottom chambers may be imaged and placed back in the incubator for later study. Chambers with different coverslip thicknesses are available to match the microscopy objective you will use. Glued coverslips can be removed from the dish using a glass bottom dish fluid (Mat Tek DCF OS 30) following the manufacture's procedure. Alternatively, glass bottom culture dishes may be constructed from acid washed coverslips using the method of Kline (Kline, 2009). Sterilize dishes with 70% ethanol and allow them to dry at the back of the cell culture hood.
Note: Mat Tek Glass bottom dishes used directly from a sterile sleeve do not require acid washing. Coat them directly with peanut lectin
Parafilm® M
Note: Perform the following procedure in a tissue culture hood and use sterile techniques.
Dissolve peanut lectin with cold sterile ddH2O in the original vial at 4 °C overnight. If the vial is small, sterilely pour the lyophilized powder into a 50 ml conical tube before adding ddH2O.
Transfer the solution from a vial to a sterile 50 ml conical tube and rinse the vial with additional cold sterile ddH2O. Keep track of the amount of water added in each step. Bring to a final concentration of 0.5 mg/ml with additional sterile ddH2O.
To coat Mat Tek glass bottom dishes, add 100 μl of 0.5 mg/ml peanut lectin to the coverslip and incubate at room temperature for 20 to 60 minutes. This peanut lectin solution can be collected and reused once to coat more coverslips.
Wash the dishes 5 times with 2 ml sterile ddH2O. Aspirate the liquid with a glass pipette connected to a vacuum trap after each wash.
To dry, place the dishes at the back of cell culture hood (most sterile environment) and tilt the lids so that they only partially cover the dish. Wait at least 1 hour or until dry.
If dishes are not used immediately, use Parafilm to wrap the dish and lid, and store at 4 °C.
Note: Peanut lectin-coated dishes can be stored for extended period, whereas peanut lectin solution is less stable. Coat a large batch of dishes with fresh peanut lectin solution and store at coated dishes at 4 °C.
Clean and dry the hemocytometer and coverglass with 70% ethanol and Kimwipes.
Dampen the mounts with a wet kimwipe or by exhaling on the hemocytometer.
Place the hemocytometer coverglass across the mounts, center, and gently press down.
Note: Hemocytometer coverglass is thicker than standard #0 or #1 coverglass and does not warp under liquid surface tension.
Insure that the cell suspension is well-mixed and pipette a 10 µl drop of cell suspension into one inlet of the hemocytometer. Wait 1 minute for cells to settle on the grid.
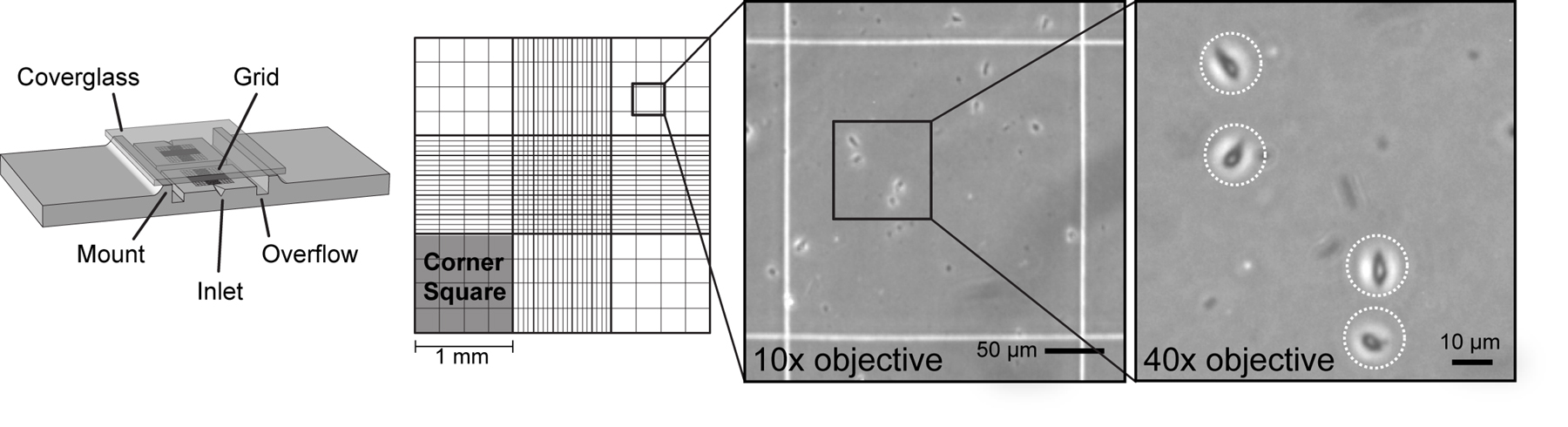
Place the hemocytometer on a phase contrast microscope and find the grid. Each corner square has an area of 1 mm2 and a depth of 0.1 mm for a total volume of 1x10–4 ml.
Count the total cells in all 4 corner-squares of the grid. The corner-squares are divided into 16 smaller squares for convenience. Intact cells will appear phase dark with a bright, white phase ring surrounding the cell (Figure 4).
Note: With practice, cells may be distinguished from debris by phase contrast. Continuously shift the focal plane up and down while counting to look for phase rings around cells. A differential interference contrast (DIC) or Hoffman modulation contrast (HMC) microscope with a 40x or 63x objective may also be used to count small cells (see Figure 14).
After counting the cells in all 4 corner-squares, divide by 4 to get the average number of cells per 1 mm2 cell area. Multiply this number by 1x104 to obtain the average number of cells per ml.
Note: For cells that lie on an edge, only count cells that cross the top or left edge of a corner square.
Disassemble the hemocytometer. Clean and dry the hemocytometer and coverglass with 70% ethanol and Kimwipes. Wrap both in lens tissue or Kimwipes and store them in a safe location.
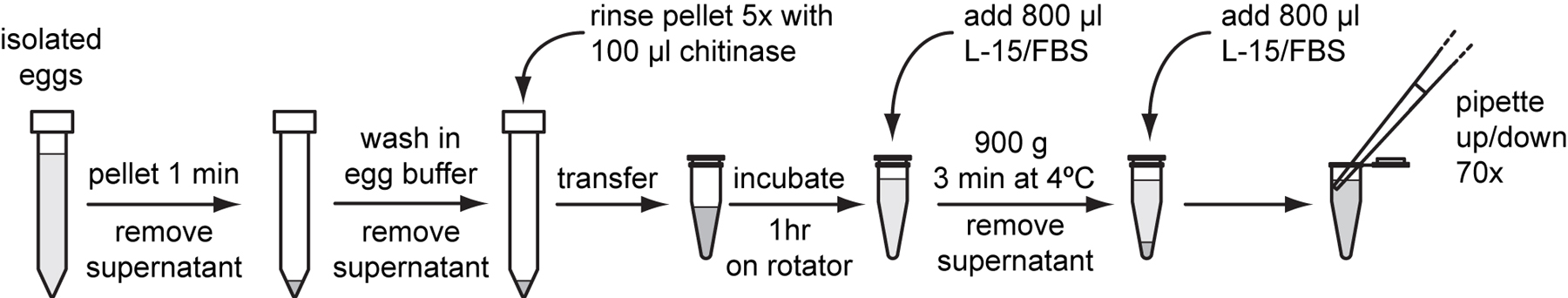
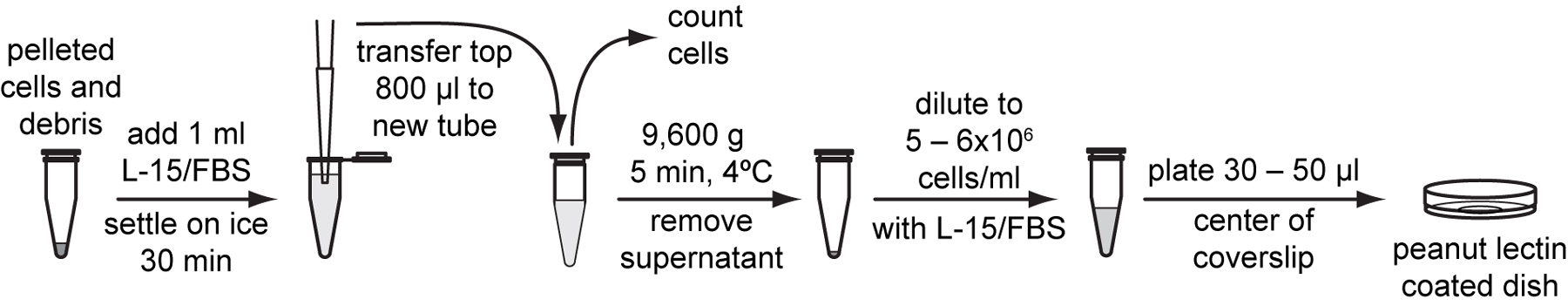
To isolate embryonic cells, gravid adults from two to three synchronized NEP-NA22 plates are dissolved to release mixed-stage embryos. Egg shells are digested with chitinase and embryos are further dissociated through pipetting. Cells are released primarily from pre-comma stage embryos. Undissociated late stage embryos and large cell clumps are separated from dissociated cells by passing the suspension through a 5 µm mesh filter. Isolated embryonic cells adhere to peanut lectin and differentiate into cells of L1 stage characteristics. The embryonic cell isolation protocol described below was developed by the Kevin Strange laboratory (Christensen et al., 2002; Strange et al., 2007). We have modified it slightly with an alternate to sucrose flotation of eggs that produces similar yields as the original protocol. We have included an extended cell filtration method that produces higher yields in our hands. See Table I below for typical yields from embryonic cell isolation.
Table I. Typical yields for embryonic and larval cell isolation.
| Cell Source | Cells per μl packed eggs or larvae | Cells per egg or larva |
|---|---|---|
| Embryo | 6.1 ± 2.9 ×104 | 3.8 ± 1.8 |
| Stage L1 | 2.7 ± 1.9 ×104 | 2.5 ± 1.7 |
| Stage L2 – L4 | 7.8 ± 1.7 ×104 | — |
Chitinase (Sigma C6137).
Sterile egg buffer.
Laboratory rotator.
3 ml syringes.
18-gauge syringe needles.
Sterile syringe filters, 5 µm pore size (Pall Corp. 4650).
Dissolve chitinase to 1 U/ml in cold sterile egg buffer.
Store 500 μl aliquots at -80 °C.
Follow steps procedures 2.2 (Propagating and synchronizing worms on NEP-NA22 plates) and 2.3 (Preparing eggs or hatched larvae) to prepare sterile eggs from 2 to 3 NEP-NA22 plates that were seeded with 20,000 hatchlings per plate.
Note: Perform the following procedure in the cell culture hood and use autoclaved or filter sterilized water and media.
Resuspend eggs in 5 ml egg buffer. Pellet eggs at 1,300 g for 1 minute in a clinical centrifuge and remove the supernatant. Pellet again at 1,300 g for 1 minute to remove as much liquid as possible. Leave approximately 100 μl of pelleted eggs.
Optional: transfer any excess eggs to another 15 ml conical tube and add 10 ml of M9. Allow to hatch overnight to seed the next round of worm culture.
Note: The embryonic cell isolation protocol described in (Christensen et al., 2002; Strange et al., 2007) separates eggs from adult carcasses by floating eggs on a layer of sucrose. This sucrose flotation step can be skipped with little difference in cell isolation outcomes if adult worms are continuously vortexed during alkaline hypochlorite digestion.
Thaw a tube of 500 µl 1 U/ml chitinase. Add 100 μl of chitinase stock to the 100 μl egg pellet and transfer to a 1.5 ml microcentrifuge tube. Rinse the original conical tube 4 times with 100 μl chitinase each time and transfer the chitinase and any suspended eggs to the same 1.5 ml tube. Rotate the reaction at room temperature for 1 hour on a laboratory rotator.
Note: Do not vortex to mix embryos or cell suspensions in the following steps.
Stop the reaction by adding 800 µl of L-15/FBS and invert the tube several times to mix.
Pellet digested embryos at 900 g for 3 minutes at 4 °C. Remove the supernatant.
Resuspend the pellet with 800 µl of L-15/FBS. Dissociate cells by pipetting the suspension up and down with a 1000 µl pipette tip against the tube side. Repeat a total of 70 times.
Monitor the progress of the digestion every 20 to 30 strokes. Touch the pipette tip to the larval suspension to pick up 1 to 2 μl of reaction, spot this sample on a glass slide, and examine under a tissue culture microscope.
When most embryos are dissociated into single cells, pellet at 900 g for 3 minutes at 4 °C. Undissociated embryos should be those at or beyond the comma stage.
Remove the supernatant and gently resuspend the pellet with 0.5 ml of L-15/FBS. Label this tube “Cells” and keep the cell suspension on ice until ready to filter.
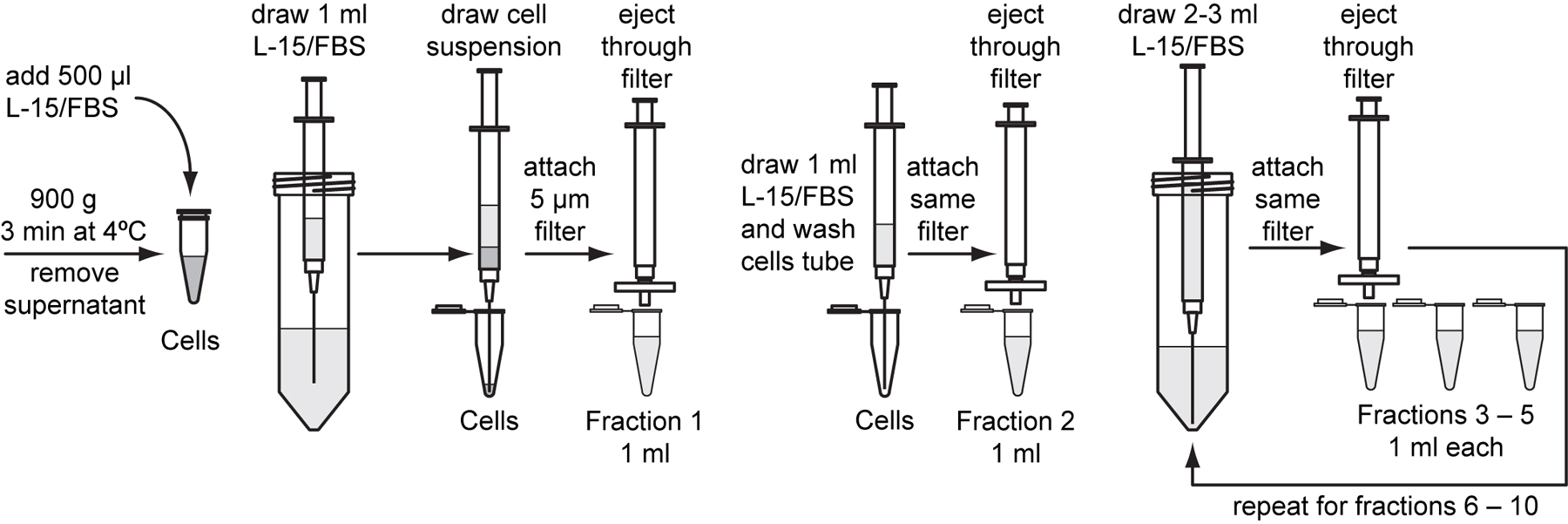
A total of 10 ml of L-15/FBS will be used to filter the suspension of dissociated embryos. Sterilely pour or pipette 12 to 15 ml of L-15/FBS into a 50 ml conical tube to use for the following wash steps.
Prepare ten 1.5 ml microcentrifuge tubes and label them “Fraction 1” through “Fraction 10”.
Fraction 1: Attach an 18-gauge needle to a 3 ml syringe. Draw 1 ml of L-15/FBS into the syringe. Then, slowly draw 0.5 ml of cell suspension into the same syringe. Be careful not to mix the two solutions.
Note: Be careful to only immerse the tip of the needle into the L-15/FBS stock solution.
Detach the needle and attach a sterile 5 µm pore-size filter to the syringe. Use the same filter for all subsequent wash steps. Gently eject the liquid through the filter into the tube marked “Fraction 1”. Approximately 0.5 ml of liquid will remain in the filter.
Fraction 2: Pipette 1 ml of L-15/FBS into the original tube marked “Cells”, swirl the tube to wash. Reattach the same 18-gauge needle to the syringe and draw the suspension into the syringe. Discard the needle and reattach the 5 µm filter. Gently eject the liquid into the tube marked “Fraction 2”.
Note: Excessive syringe pressure will damage the cells, while low pressure reduces cell yields in the first two fractions. Collecting additional fractions 3 through 10 and assaying cell yields can compensate for variations in syringe pressure between users and different brands of syringe and filter. We recommend that each user optimize the number of collected fractions for syringe, pressure, and filter. Fewer fractions may be collected once repeatability is established.
Fractions 3-5: Attach a new 18-gauge needle to the syringe and draw 3 ml of L-15/FBS into the syringe. Remove the needle and reattach the 5 µm filter. Gently eject the 1 ml at a time into the tubes marked “Fraction 3” through “Fraction 5”.
Fractions 6-8: Reattach the 18-gauge needle to the syringe and draw 3 ml of L-15/FBS into the syringe. Remove the needle and reattach the 5 µm filter. Gently eject the 1 ml at a time into the tubes marked “Fraction 6” through “Fraction 8”.
Fractions 9-10: Reattach the 18-gauge needle to the syringe and draw 2 ml of L-15/FBS into the syringe. Remove the needle and reattach the 5 µm filter. Gently eject the 1 ml at a time into the tubes marked “Fraction 9” and “Fraction 10”.
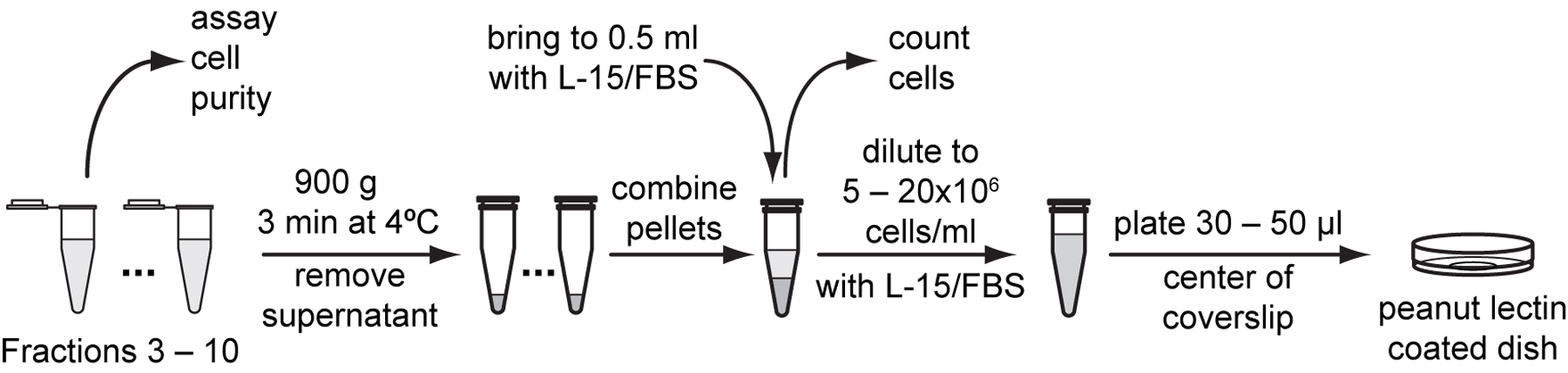
To assay cell purity, sample 5 μl of cell suspension from each fraction, place onto a microscope slide and observe under a cell culture microscope. Keep the fractions with most cells and little debris.
Note: The expected outcomes of the filtration are: fraction 1-3: milky with very small pieces of particles (discard); fraction 4: a few cells mixed with some debris (discard); fractions 5-10: several cells mixed with little debris (keep).
Pellet tubes with the most pure cells in a microcentrifuge at 900 g for 3 minutes at 4 °C. Remove all but the last 10 to 20 µl supernatant.
Gently resuspend cell pellet with the residual medium left in the tube. Combine cell suspensions into one tube and resuspend to approximately 500 µl with L-15/FBS.
Use a hemocytometer to count cell density in 10 µl of cell suspension and calculate total number of cells in the suspension (Protocol 4.5).
Resuspend cells with L-15/FBS to a final density of 5x106 to 20x106 cells /ml.
Plate 30-50 µl of cell suspension onto the center of peanut lectin-coated glass bottom dishes. Place dishes in a plastic container with a gas exchange hole and humidified with Kimwipes wetted with ddH2O. Allow cells to adhere overnight in a 20 to 23 °C incubator.
The next day, pre-warm L-15/FBS to room temperature.
Wash off unbound cells and larval debris once with 1 ml of L-15/FBS. Aspirate or pipette to remove the media.
Add 0.5 to 1 ml of fresh L-15/FBS to the dish and return to the incubator.
Freshly synchronized L1 larvae from one to three NEP-NA22 plates are used for L1 cell isolation. Synchronized CeHR worm culture is used for later stage larval cell isolation. For all stage larval cell isolation, a brief chemical treatment (SDS-DTT) is used to pre-sensitize worm cuticle. The cuticle is then enzymatically digested with pronase E and cells are released with mechanical disruption (pipetting). SDS-DTT and pronase E treatment times have been optimized by stage to reduce cell damage and promote cell survival. See Table I in section 5 (Embryonic Cells) for typical yields from larval cell isolation.
1 M HEPES, pH 8.0. Make a 1M solution in ddH2O and bring to pH 8.0.
10% Sodium dodecyl sulfate (SDS). Make a 10% (w/v) solution in ddH2O and store at 4 °C.
1 M Dithiothreitol (DTT). Make 1 M stocks in room temperature ddH2O, aliquot, and store at −20 °C. As a reducing agent, DTT is the most sensitive chemical in the larval dissociation protocol and a likely cause of poor larval dissociation. Purchase small bottles of DTT (5 to 10 g) and store at −20 °C in a box with desiccant. Wrap the bottle top with Parafilm after each use. DTT may precipitate in cold water. When thawing either DTT stock or SDS-DTT stocks, warm the aliquot in your hand and vortex the aliquot until clear.
60% Sucrose. Make a 60% (w/v) solution in ddH2O and autoclave.
Egg buffer.
L-15/FBS media.
15 mg/ml Pronase E (Sigma P8811) in egg buffer.
Assemble the following components:
| Component and final concentration | Stock | 10.25 ml |
|---|---|---|
| 20 mM HEPES pH 8.0 | 1 M | 0.2 ml |
| 0.25% SDS | 10% | 0.25 ml |
| 200 mM DTT | 1 M | 2 ml |
| 3% sucrose | 60% | 0.5 ml |
| ddH2O | – | 7.3 ml |
In a tissue culture hood, sterilize SDS-DTT solution using a 0.2 μm syringe filter. Store 200 μl aliquots at −20 °C.
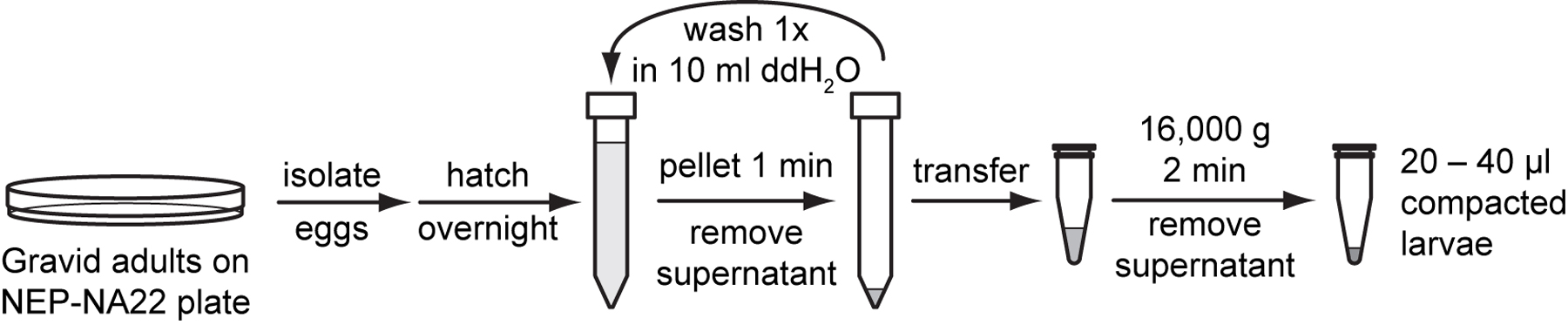
Seed 2 to 3 NEP-NA22 plates with 20,000 synchronized L1 per plate (see protocol 2.2, Propagating and synchronizing worms on NEP-NA22 plates).
Allow worms to reach gravid adult stage.
Use the same 10 ml of ddH2O to wash gravid adults from each plate and dispense the combined worm suspension into one 15 ml conical tube.
Centrifuge at 1,300 g for 1 minute in a clinical centrifuge. Remove water from the top of the supernatant to leave 7 ml of total volume.
Note: Perform the following procedure in the cell culture hood and use filter sterilized or autoclaved water and media.
Use protocol 2.3 (Preparing eggs or hatched larvae) to obtain eggs in M9 buffer. Allow the eggs to hatch overnight on a laboratory rotator.
Note: Wipe pipettors with 70% ethanol after possible contact with bacteria during egg isolation.
The next day, pre-warm L-15/FBS and egg buffer to room temperature.
Note: For the following steps, do not vortex to mix larvae or cell suspensions.
Pellet synchronized L1 by centrifugation at 1,300 g for 1 minute in a clinical centrifuge. Remove M9.
Resuspend the larval pellet with 10 ml of sterile ddH2O and gentle pipetting. Pellet worms again by centrifugation (1,300 g for 1 minute). Sterilely pour off the supernatant, leaving roughly 1 ml total of pellet and supernatant.
Prepare a 1.5 ml microcentrifuge tube by filling it with 40 µl of sterile ddH2O and marking the water level on the outside of the tube.
Transfer pelleted larvae to the prepared microcentrifuge tube. Pellet larvae at 16,000 g for 2 minutes in a microcentrifuge and carefully remove the residual ddH2O with a 200 µl pipette tip to leave a compact pellet.
Use 20 to 40 μl of compacted L1 for isolation. If pellet is larger than 40 µl, resuspend the pellet in a small amount of ddH2O and remove some of the suspended larvae. Re-pellet the suspension and check the level of packed larvae before beginning.
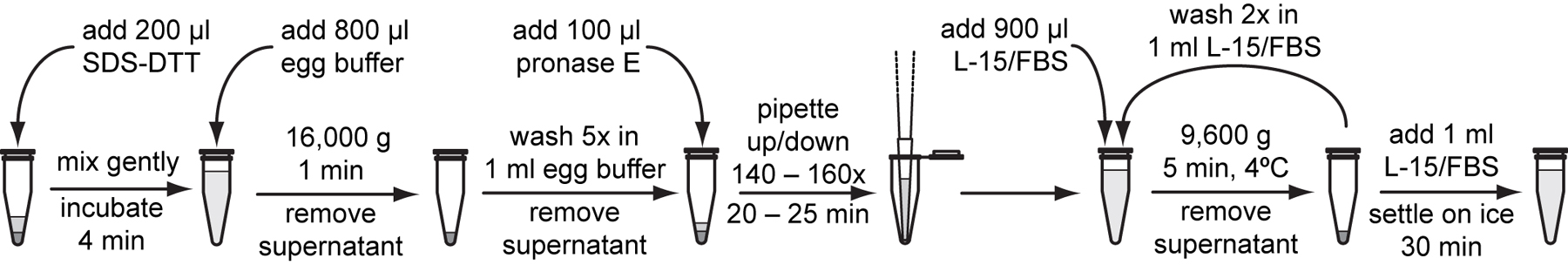
Add 200 μl freshly thawed SDS-DTT solution to the larval pellet and gently flick the bottom of the tube with the fingertip until mixed. Incubate for exactly 2 minutes at room temperature.
Note: DTT precipitates at low temperature. Warm the SDS-DTT aliquot in the hand to thaw and vortex until it is clear of precipitates before using.
Immediately after the SDS-DTT treatment, add 800 μl egg buffer to the reaction. Flick gently to mix.
Pellet larvae at 16,000 g for 1 minute. Remove the supernatant and wash 5 more times with 1 ml egg buffer per wash. Resuspend pellets by flicking the tube for each wash.
Note: It is critical to perform steps 12–14 quickly, as L1 survival drops dramatically if incubated in SDS-DTT for more than 2 minutes.
Add 100 μl freshly thawed 15 mg/ml pronase E to washed larval pellet, flick to mix, and incubate at room temperature for 7 to 9 minutes.
Pipette the larvae suspension with a 200 µl tip during the digestion. Adjust the pipetting volume to the approximate volume of the suspended pellet. Slowly pull suspended larvae into the pipette tip. Then, press down to force the pipette tip against the bottom of the microcentrifuge tube and slowly eject the contents. Take approximately 6 to 8 seconds for the downward stroke. Repeat the cycle of up/down strokes a total of 60 to 90 times during the incubation.
Monitor the progress of the digestion every 2 to 3 minutes (every 20-30 strokes). Touch the pipette tip to the larval suspension to pick up 1 to 2 μl of reaction, spot this sample on a glass slide, and examine under a tissue culture microscope.
Note: The SDS-DTT treatment and Protease digestion are kept short to maximize cell yields and viability, not larval dissociation. L1 stage larvae are particularly resistant to protease digestion and will yield fewer cells than will later stage larvae. Expect several live, undissociated, larvae after digestion. These will be separated from cells in steps 20-21.
Stop the reaction by diluting with 900 μl of L-15/FBS.
Centrifuge the digested larvae at 9,600 g for 5 minutes at 4 °C. Remove supernatant and wash 2 more times with 1 ml L-15/FBS per wash. Pipette the suspension to mix each time.
To remove most of the undigested larvae and debris, resuspend the pellet with 1 ml L-15/FBS and settle the suspension on ice for 30 minutes.
Gently transfer the top 800 μl cell suspension devoid of large larval debris to a new tube.
Use a hemocytometer to count cell density in 10 µl of cell suspension and calculate total number of cells in the suspension (Protocol 4.5).
Note: Count only large and medium sized cells. Small cells are difficult to distinguish from debris.
Centrifuge the cell suspension at 9,600 g for 5 minutes at 4 °C.
Note: The embryonic cell isolation protocol pellets cells at a lower relative centrifugal force of 900 g. The higher speeds used here slightly increase the yield of small cells without introducing cellular damage.
Resuspend cells with L-15/FBS to a final density of 5x106 to 6x106 cells /ml according to the cell number calculated in step 22.
Plate 30 to 50 µl of cell suspension onto the center of peanut lectin-coated glass bottom dishes. Place dishes in a plastic container with a gas exchange hole and humidified with Kimwipes wetted with ddH2O. Allow the cells to adhere overnight in a 20 to 23 °C incubator.
The next day, pre-warm L-15/FBS to room temperature.
Wash off unbound cells and larval debris once with 1 ml of L-15/FBS. Aspirate or pipette to remove the media.
Note: Some cells require more than 24 hours to adhere (David Miller, personal communication). Experiment with adhesion times before washing debris and unattached cells from the coverslip.
Add 0.5 to 1 ml of fresh L-15/FBS to the dish and return to the incubator.
Note: Perform the following procedure in the cell culture hood and use filter sterilized or autoclaved water and media. Do not vortex to mix larvae or cell suspensions.
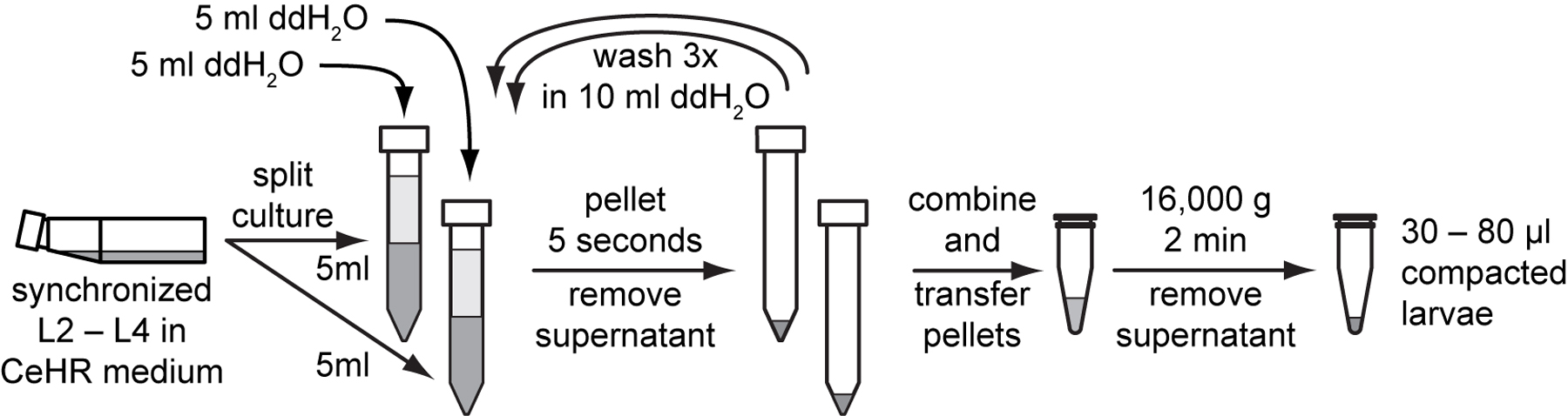
Grow larvae to the desired stage in CeHR medium. Harvest 1 to 2 flasks of worms for the cell preparation.
For each flask, split the 10 ml of CeHR larvae culture into two 15 ml conical tubes. Add 5 ml of sterile ddH2O to each 5 ml of larvae culture and mix by inverting the tubes several times. Place tubes in a clinical centrifuge and set the speed to 1,300 g. Start the centrifuge, wait 5 seconds, and stop the centrifugation.
Note: Although the centrifuge will not reach its maximum speed, a brief, five-second centrifugation separates larvae from large particulate matter in the CeHR medium.
Remove the supernatant with a pipette and add 10 ml of sterile ddH2O to each tube and invert the tube several times to resuspend the larvae. Pellet the larvae again for 5 seconds in a clinical centrifuge.
Repeat step 3 two more times for a total of three ddH2O washes.
Remove most of the supernatant, leaving approximately 1 ml of total pellet and supernatant.
Prepare a 1.5 ml microcentrifuge tube by filling it with 80 µl of sterile ddH2O and marking the water level on the outside of the tube.
Transfer pelleted larvae to the prepared microcentrifuge tube. Pellet larvae at 16,000 g for 2 minutes in a microcentrifuge and carefully remove the residual ddH2O with a 200 µl pipette tip to leave a compact pellet.
Use 30 to 80 μl of compacted L2–L4 for isolation. If pellet is larger than 80 µl, resuspend the pellet in a small amount of ddH2O and remove some of the suspended larvae. Re-pellet the suspension and check the level of packed larvae before beginning.
Add 200 μl freshly thawed SDS-DTT solution to the larval pellet and gently flick the bottom of the tube with the fingertip until mixed. Incubate for exactly 4 minutes at room temperature.
Note: DTT precipitates at low temperature. Vortex the thawed SDS-DTT until it is clear of precipitates before using.
Immediately after the SDS-DTT treatment, add 800 μl egg buffer to the reaction. Flick gently to mix.
Pellet larvae at 16,000 g for 1 minute. Remove the supernatant and wash 5 more times with 1 ml egg buffer per wash. Resuspend pellets by flicking the tube for each wash.
Note: It is critical to perform steps 9 – 11 quickly, as larval survival drops dramatically if incubated in SDS-DTT for more than 4 minutes.
Add 100 μl freshly thawed 15 mg/ml pronase E to washed larval pellet, flick to mix, and incubate at room temperature for 20 to 25 minutes.
Pipette the larvae suspension with a 200 µl tip during the digestion. Adjust the pipetting volume to the approximate volume of the suspended pellet. Slowly pull suspended larvae into the pipette tip. Then, press down to force the pipette tip against the bottom of the microcentrifuge tube and slowly eject the contents. Take approximately 6 to 8 seconds for the downward stroke. Repeat the cycle of up/down strokes a total of 140 to 160 times during the incubation.
Monitor the progress of the digestion every 3 to 5 minutes (every 30-50 strokes). Touch the pipette tip to the larval suspension to pick up 1 to 2 µl of reaction, spot this sample on a glass slide, and examine under a tissue culture microscope.
Note: The SDS-DTT treatment and Protease digestion are kept short to maximize cell yields and viability, not larval dissociation. Expect several live, undissociated, larvae after digestion. These will be separated from cells in steps 17-18.
Stop the reaction by diluting with 900 μl of L-15/FBS.
Centrifuge the digested larvae at 9,600 g for 5 minutes at 4 °C. Remove supernatant and wash 2 more times with 1 ml L-15/FBS per wash. Flick the tube gently to mix each time.
To remove most of the undigested larvae and debris, resuspend the pellet with 1 ml L-15/FBS and settle the suspension on ice for 30 minutes.
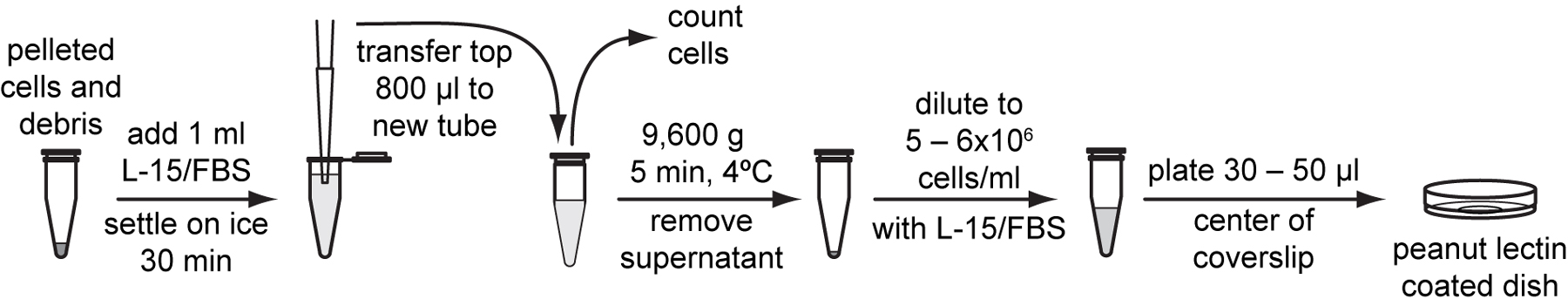
Gently transfer the top 800 μl cell suspension devoid of large larval debris to a new tube.
Use a hemocytometer to count cell density in 10 µl of cell suspension and calculate total number of cells in the suspension (Protocol 4.5).
Note: Count only large and medium sized cells. Small cells are difficult to distinguish from debris.
Centrifuge the cell suspension at 9,600 g for 5 minutes at 4 °C.
Resuspend cells with L-15/FBS to a final density of 5x106 to 6x106 cells /ml according to the cell number calculated in step 19.
Plate 30 to 50 µl of cell suspension onto the center of peanut lectin-coated glass bottom dishes. Place dishes in a plastic container with a gas exchange hole and humidified with Kimwipes wetted with ddH2O. Allow the cells to adhere overnight in a 20 to 23 °C incubator.
The next day, pre-warm L-15/FBS to room temperature.
Wash off unbound cells and larval debris once with 1 ml of L-15/FBS. Aspirate or pipette to remove the media.
Note: Some cells require more than 24 hours to adhere (David Miller, personal communication). Experiment with adhesion times before washing debris and unattached cells from the coverslip.
Add 0.5 to 1 ml of fresh L-15/FBS to the dish and return to the incubator.
Isolated cells provide stage-specific developmental “snapshots” of cell-autonomous behaviors. Cells isolated from different stages vary in their morphologies (Figure 14). Both cell size and the fraction of multinucleate cells increase in isolates from L2 to L4 stages (Zhang et al., 2011). This increase mirrors cell growth and formation of syncytia in the maturing larvae. L2 to L4 cells also show a richer variety of morphologies than do embryonic (Christensen et al., 2002) and L1 stage cells (Zhang et al., 2011), where either muscle (L1) or muscle and neurons (embryonic) predominate in culture.
|
Figure 14. DIC micrographs of one-day culture of cells from embryos and L1 through L4 stage larvae. Several cellular morphologies can be seen including muscle cells (white arrowhead), neurons (white arrow), and cells with large protrusions (large white arrows). L4 stage cell morphologies include multiple nuclei (black arrow) and cleavage furrows (black arrowhead).
Cellular cross-talk can influence cellular differentiation in mixed cell cultures (Moldaver and Yegorov, 2009; Portela et al., 2010; Lin and Talbot, 2011). To compensate, mixed cell populations can be reduced to single cell types through fluorescence-activated cell sorting (FACS, or flow cytometry). Both high and low abundant embryonic cells from GFP marker strains can be isolated, sorted, and cultured for several days (Christensen et al., 2002; Strange et al., 2007). Larval cells can be sorted and enriched using similar methods (D. Miller, personal communication). Thus, mono-cell culture provides a finely controlled environment to study cell-autonomous differentiation and behavior.
When combined with FACS, cell isolation can also be used to profile individual embryonic or larval cells in a near-in vivo state. Cell isolation can be performed in approximately two hours and modern flow cytometry instruments can sort through thousands of cells per second. Thus, cells can be isolated and ready for experiments within three hours of separation from their in vivo niches. For example, the transcriptome (Fox et al., 2005; Von Stetina et al., 2007; Spencer et al., 2011) or proteome (Husson et al., 2009; Shim and Paik, 2010) of specific cell types can be profiled with microarrays or mass spectrometry of FACS sorted cells. By isolating embryonic through L4 stage cells, cell linages can be followed over the course of development. More traditional techniques such as co-immunoprecipitation and Western blots can be used to follow cell-specific protein-protein interactions. In each case, the extension of cell isolation from embryos to larvae has the potential to substantially further the study of physiological, cellular, and molecular phenomena at the single cell level.
Co-culture of two different cell populations is increasingly being used to study specific cell-cell interactions (Casbas-Hernandez et al., 2011; Kaji et al., 2011). We envision that sorting and co-culture of two different cell populations will provide a unique opportunity to study specific non-cell-autonomous interactions in nematodes. For example, cells providing morphogens or trophic factors could be co-cultured with appropriate cells types to test models of stage-specific vulval development (Fisher et al., 2005; Sternberg, 2005), neuronal guidance cues (Killeen and Sybingco, 2008; Quinn and Wadsworth, 2008), or stem cell niches (Waters and Reinke, 2011; Hajduskova et al., 2012). Stage-specific signal generation or response could be studied by varying the isolation time of one of the two cell populations.
Isolated cells spread on two-dimensional substrates also can be used to observe sub-cellular dynamics with greater precision than available in vivo. For example, total internal reflection fluorescence (TIRF) microscopy can follow fluorescent “speckles” in cells containing one to a few fluorophores (Danuser and Waterman-Storer, 2006). TIRF microscopy limits excitation to thin “evanescent wave” at the coverslip-buffer interface and substantially reduces both background fluorescence and photobleaching. This 100–200 nm thick, exponentially decaying excitation layer (Axelrod, 2001) only weakly penetrates the 100–150 nm thick cuticle of L1 worms and cannot penetrate the 260–350 nm thick cuticle of L4 worms (Cox et al., 1981) or the 300–400 nm thick eggshell (Johnston and Dennis, 2012). Consequently, in vivo TIRF microscopy in C. elegans has been limited to one-cell and two-cell embryos (Hyenne et al., 2012). In contrast, TIRF microscopy performs optimally on isolated cells on planar substrates. TIRF microscopy of isolated cells can be used to track the cytoskeleton (Figure 15) (Hu et al., 2007; Nemethova et al., 2008), adhesion complexes (Adams et al., 2004; Pasapera et al., 2010), or endocytosis/exocytosis (Sund et al., 1999; Rohrbach, 2000; Lin et al., 2010) with great detail.
|
Figure 15. Actin filaments in isolated embryonic M-lineage cells. (A) Actin filaments were labeled with the actin binding domain of Utrophin (Burkel et al., 2007) linked to mEGFP. Live cells from this hlh-8::mEGFP-UtrCH marker strain were imaged with total internal reflection fluorescence (TIRF) microscopy. (B) Time-lapse TIRF micrograph of actin filaments in an isolated M-lineage cell. Times shown in min:sec.
Large-scale embryonic cell culture was originally applied to electrophysiology (Christensen et al., 2002). Although recording of voltages and ion currents through thin glass pipettes can be done in individual filleted animals (Goodman et al., 1998; Richmond and Jorgensen, 1999), the filleting technique is extremely demanding. In contrast, isolated neurons can be produced in abundance and followed for two weeks in culture. Isolated cells are easy to access with patch pipettes for single channel to whole cell recording (see WormMethods chapter, Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses: doi/10.1895/wormbook.1.122.1, http://www.wormbook.org). The protocols outlined here will extend isolated-cell electrophysiology to postembryonic lineages.
Neuronal genesis in vivo is complete by stage L2 (Sulston and Horvitz, 1977), but neurons undergo dramatic post-mitotic changes over the course of development (White et al., 1986; Varshney et al., 2011). Cells lose most processes during the isolation step, but neurons from all stages regenerate dendrites and long axons on peanut lectin coated glass over the course of several days (Zhang et al., 2011). Thus, neuronal isolation from later larval stages could be used to partially recapitulate the dramatic axonal growth and guidance in vitro that occurs during larval development in vivo. Co-culture of neurons with target cells or gradients of trophic factors could be used to study the specificity of axonal targeting ex vivo.
We thank Diya Banerjee for helpful discussions and technical advice. This work was supported by a Burroughs-Wellcome Career Award at the Scientific Interface to JRK and a Virginia Tech Institute for Critical Technology and Applied Science Doctoral Fellowship to SZ.
|
Figure 16. Major cell culture equipment and supplies include: (1) class II flow hood, (2) incubator with (3) chilled water supply, (4) refrigerator/freezer, (5) vacuum trap, (6) box for sterile glass pipettes, (7) pipette tips, (8) microscope, (9) osmometer, (10) clinical centrifuge, (11) refrigerated microcentrifuge, (12) Pipet-Aid, (13) spray bottle with 70% ethanol.
Class II laminar flow hood. A class II microbiological safety cabinet passes all outside air through a HEPA filtered inlet and thus provides a sterile working area free from dust and microbial contamination. Class II cabinets also contain a UV light source to maintain sterility inside the cabinet when the cabinet is not in use. In a typical class II cabinet, outside air is pulled in through a grating just below the sash. Outside air is kept separate from the working area and channeled toward a HEPA filter at the top of the cabinet. Filtered air is then blown downward into the working environment. A grating at the back of the work surface recirculates some of this air through the HEPA filter, while the rest escapes to the outside under the sash. Slight positive pressure inside of the working area creates an invisible curtain, sandwiched between the inlet grating and the bottom of the sash, that separates the outside environment from the filtered air inside the working environment. The working surface just beyond the inlet grating can be maintained as sterile. Class II cabinets are available with or without an adjustable sash. Either is sufficient for nematode cell culture. Ideally, the cabinet should be placed in a dedicated tissue culture room. If a dedicated room is not available, place the cabinet in a low-traffic area of the laboratory.
Note: A class I fume hood used for working with volatile or toxic chemicals is not sufficient for cell culture. These cabinets only filter exhaust air and are designed to protect the user from contamination, not the working area inside of the cabinet.
Laboratory incubator for 20 °C (optional). Nematode cells should be kept in a sterile environment at a constant temperature and away from any plates containing bacteria. A CO2 supply is not necessary to maintain media pH. Many standard 37 °C tissue culture incubators can be converted to a 20 °C incubator by closing the CO2 valve and attaching a chilled circulating bath filled with tap or distilled water (not deionized) to the incubator's water jacket. If an incubator is not available, dishes with cultured cells can be kept at room temperature and away from bacteria plates in sterile, plastic containers. (Suggested model: Nuaire NU-4750 incubator and Julabo F12 water bath)
Laboratory refrigerator/freezer combination. A separate refrigerator/freezer unit for tissue culture media and ingredients is recommended to minimize contamination.
Two-stage vacuum trap for aspiration. Construct an aspirator for the culture hood using two Buchner or Erlenmeyer flasks, two 1-hole or 2-hole rubber stoppers, glass Pasteur pipettes or glass tubes, and Tygon® tubing as shown in Figure 16 above. Use a 1000 ml or larger flask for the first stage trap. Connect the second stage to a vacuum supply or pump through a vacuum filter to protect the pump from residual water vapor.
Stainless steel pipette sterilization boxes. Obtain at least two sterilization boxes and keep one autoclaved box of pipettes in the culture hood at all times. (Suggested model: Fisher Scientific 03-475-5 rectangular box for 9 inch Pasteur pipettes)
Set of pipettes for flow hood. Keep a separate set of autoclavable 2–20 µl, 20–200 µl, and 100–1000 µl pipettes to be used only for cell culture. Autoclave the pipettes according to the manufacturer's instructions. Autoclave again every one to two months or in case of contamination.
Tissue culture microscope. An inverted, phase contrast microscope with 10x, 20x, and 40x phase-contrast objectives and a long working distance condenser should be used for observing the extent of cell extraction, for counting cells, and for cell maintenance.
Stereo dissecting microscope. A standard stereo microscope used for nematode maintenance and counting.
High-resolution epi-fluorescence/DIC inverted microscope. An inverted microscope with objective lens below the specimen may be used for observing the behavior of cultured cells. We recommend epi-fluorescence illumination for locating GFP expressing cells and differential interference contrast (DIC) optics for observing cellular behavior. Alternatively, an upright microscope with a water-immersion lens may be used for imaging cultured cells plated on glass-bottom petri dishes.
Osmometer. Either a vapor pressure osmometer or freezing point depression osmometer can be used to estimate and adjust cell culture media osmolarity before filtration. Adjustment is done infrequently, and a shared osmometer or one located in a different building is sufficient. (Suggested model: Wescor Vapro 5520 or 5600)
Clinical centrifuge. Either a fixed-angle or swinging bucket centrifuge for 15 ml conical tubes with a maximum speed of 1,300 g can be used for pelleting nematodes and floating eggs on sucrose. (Suggested model: Fisher Scientific 228 Benchtop Centrifuge)
Refrigerated centrifuge. Use a refrigerated centrifuge with maximum speed of ~16,000 g for 1.5 ml tubes. If a refrigerated microcentrifuge is not available, keep one non-refrigerated microcentrifuge in a cold room or laboratory refrigerator and one non-refrigerated microcentrifuge at room temperature on a nearby bench-top.
Portable serological Pipet-Aid® (Drummond Scientific) for flow hood. An electric Pipet-Aid with aspiration and dispensing buttons and a sterile inlet filter. This Pipet-Aid should be used only for cell culture. Keep a supply of spare filters to replace wetted or contaminated filters.
Hemocytometer. Obtain a standard, four-quadrant glass hemocytometer slide for counting cells. Note that hemocytometers use non-standard, 0.5 mm thick coverslips. These thick coverslips do not bend under liquid surface tension and insure a consistent volume for cell counting. Hemocytometer coverslips can be rinsed and reused indefinitely, but keep at least one extra coverslip at hand in case of breakage.
Glass bottles. Cell culture glassware should be kept separate from other laboratory glassware to limit contamination from residual detergents, salts, lipids, and proteins. Obtain a new set of 500 and 1000 ml Pyrex or borosilicate glass bottles with GL45 (Corning®) caps and label them as “Cell Culture Only” with a paint pen. Wash all new glassware in a detergent compatible with tissue culture such as Decon® 90 or 7X® (MP Biochemicals) to remove factory and storage contaminants. Fill the bottle with deionized water and autoclave on a liquid cycle with the tops loose. Wash and rinse bottles once more. Fill with deionized water, and autoclave again. Thereafter, do not use detergents in tissue culture glassware or limit their use.
Reusable filter holders (Nalgene DS0320-5045). Nutrient-rich culture media must be sterile filtered for storage and use. Reusable, autoclavable bottle-top filter holders are less expensive over time than disposable filter bottles. Obtain two to four 500 ml filter holders that screw onto standard GL45 threaded bottles. Alternatively, disposable bottle top filter sterilizers may be used to save time.
Laboratory rotator. A slow tube rotator or vertical carousel may be used to hatch nematode eggs.
Incubated orbital shaker. A variable speed incubated shaker is used to grow nematodes in CeHR axenic media at 22 °C. A non-sterile shaker/incubator may be used if nematodes are grown in T25 flasks with vented 0.2 µm filter caps for gas exchange. Alternatively, an open-air shaker may be used if larval stage is regularly monitored during growth in CeHR media.
Vortex mixer. General-purpose laboratory variable-speed mixer for 1.5, 15, and 50 ml conical tubes.
Serological pipettes (10 ml, 5 ml). Sterile, graduated, plastic pipettes for tissue culture come individually wrapped and have a cotton plug at one end to prevent wetting of the Pipet-Aid filter. Keep several in a bin or rack very close to the culture hood to minimize arm and body motion that might waft unfiltered air into the hood. Always store pipettes in the same orientation to prevent accidentally opening them from the wrong end.
Sterile pipette tips. Autoclave racks of pipette tips and keep at least one box of each size in the culture hood at all times. Do not remove from the hood after opening.
Glass Pasteur pipettes.
Sterile syringes (10 ml, 3 ml). Use individually wrapped syringes.
Sterile syringe filters, 0.22 µm pore size, either PVDF or PES. Use individually wrapped filters for sterilizing small quantities of liquids.
Sterile syringe filters, 5 µm pore size. Large pore filters are used for embryonic cell isolation. They are not required for larval stage cell isolation (Suggested model: Pall Corp. 4650).
Sterile membrane filters, 0.22 µm pore size, 47 mm diameter. Membrane filter diameter should match the diameter of the reusable bottle-top filter housing. (Suggested Model: Millipore GSWG047S6). Membrane filters are not required if disposable bottle-top filters are used.
Disposable vacuum filters, 0.2 – 0.22 µm pore size, (500 ml). Disposable filters with integrated plastic receiving flasks are available from a number of manufacturers. They are a convenient substitute for reusable bottle-top filters.
Sterile conical tubes (15 ml, 50 ml). Purchase either in bulk or in disposable Styrofoam racks. Open the plastic sleeve only at one end and only in the hood. The plastic sleeve may be discarded if the rack is left permanently in the hood. If the rack needs to be removed from the hood to make room, roll the open end of the plastic sleeve closed and affix with tape before removing from the sterile environment.
Spray bottle with 70% ethanol. Do not use pure ethanol (190-200 proof), as it is not as effective at killing mold and bacteria as 70% ethanol.
Kimwipes. Either large or small Kimwipes (Kimberly-Clark) or equivalent low lint wipe should be kept near but not within the cell culture hood. An acrylic Kimwipe box holder affixed to the culture hood above the sash is a convenient location.
Powder free gloves. Powdered residue from the manufacturing process will contaminate culture media and cells. Keep powder-free, residue-free latex, vinyl, or nitrile gloves near the culture hood, but not within it.
1.5 ml microcentrifuge tubes with snap caps.
T25 tissue culture flasks, uncoated, with vented caps. These 25 cm2 surface area canted neck flasks are used for growing larvae in axenic growth media. The vented cap contains a 0.2 µm filter for sterile gas exchange. They are not required for embryonic or L1 stage cell culture.
Osmometer calibration standards and sample discs. Calibration standards come in sealed glass ampoules that should be used immediately and discarded. For a vapor pressure osmometer, consult the manufacturer for the appropriate disposable sample disc filters. (Suggested models: Wescor Opti-Mole 100, 290, and 1000 mmol/kg standards and Wescor sample discs).
Sodium Hypochlorite (household bleach). Only use bleach to sterilize glass or plastic. Halides such as bleach corrode metal. Do not use bleach to sterilize stainless steel or any other metal surfaces.
Because C. elegans do not harbor any known human, mouse, or primate infectious agents, nematode cell culture requires only Bio-Safety Level 1 (BSL-1). Nevertheless, maintaining cells in culture requires attention to sterility beyond that required by the aseptic techniques as practiced by most laboratories that maintain nematodes. Cell culture media containing fetal bovine serum (FBS) are rich in nutrients and highly prone to bacterial and fungal contamination, even in the presence of limited antibiotics. Numerous resources on sterile technique for cell culture are available and include books (Barker, 2004), technical handbooks from major media suppliers (GIBCO), and online videos (YouTube). The equipment, supplies, and rules listed below are the minimum needed to extend common C. elegans sterile techniques to nematode cell culture.
Note: Open flames are no longer recommended for use inside of a class II laminar flow hood (Centers for Disease Control and Prevention, CDC). An open flame presents a fire hazard when used with 70% ethanol. Furthermore, heat disrupts the airflow pattern within the cabinet and can damage the HEPA filter.
Tie back long hair and remove any rings, watches, or bracelets. Wash both hands and wrists thoroughly with antibacterial soap.
Don a laboratory coat and powder free latex or vinyl gloves after washing and drying.
Spray gloves with 70% ethanol and dry with Kimwipes.
Change gloves after leaving or reentering the culture area.
Note: Do not touch your face, hair, pockets, clothes, or phone while working in the hood. Turn off your cell phone to avoid distractions. If you accidentally touch any potentially contaminated surface, decontaminate gloves with ethanol or change gloves depending on the extent of the potential contamination.
Before working in the culture hood, turn off the UV lamp, turn on the blower, and raise the sash to the working mark if the sash is adjustable.
Organize the work surface so that the area immediately in front is clear. Thoroughly spray the work surface inside the hood with 70% ethanol and wipe with Kimwipes.
Note: Disinfect often. When leaving the culture hood for more than 30 minutes or when changing operators, disinfect the work surface with 70% ethanol.
Disinfect items such as Pipet-Aids, bottles, and tubes with 70% ethanol and wipe with Kimwipes before placing them in the culture hood.
Move permanent residents of the hood such as sterile pipette boxes and racks for conical tubes to either side of the work surface when not in use.
Note: HEPA filters remove most particulate matter from air, but a small fraction of a percent of dust, bacteria, spores, etc. pass through. This percentage increases with filter age, so have the culture hood checked and certified yearly. Though the chances of contaminating your cultures and media in the hood are much slimmer than on a bench, they are not zero. When training, it may be helpful to visualize a constant rain of contaminating particles inside of the hood.
Organize the work surface of the culture hood. Move any permanent residents of the hood such as racks and pipette tip boxes to either side of the work surface.
Disinfect the culture hood work surface with 70% ethanol.
Disinfect the vacuum trap by aspirating a small amount of household bleach directly into the Tygon tubing. Follow with distilled water. Spray and wipe the tubing end with 70% ethanol. Discard liquid in the trap according to your institution's safety policies.
Lower the sash to its closed position (if adjustable), turn off the blower, and turn on the UV lamp.
Organize and disinfect any other open bench tops in the cell culture area with 70% ethanol.
If any at-hand stocks such as serological pipettes or gloves are low, refill the drawer or container. Properly dispose of sharps containers and trash when full.
Note: Be mindful of coworkers. Unlike your personal laboratory bench, cell culture areas are usually shared resources. Sloppiness can ruin your colleagues’ experiments. When you are done, insure that the cell culture area is tidy, organized, and clean. Remember that cleanup is an essential part of any experiment!
Disinfect any bottles and containers before placing in the culture hood. Spray the outsides and bottoms with 70% ethanol and wipe down with Kimwipes.
Reorganize containers to minimize motion during each step in a protocol. For example, if you are right handed, place pipette tip boxes to the right of any media bottles you are working with. Work slowly and deliberately.
Note: Only open sterile containers in the culture hood. If a container must be left open for an extended period (i.e., UV sterilization), place it at the back of the culture hood to minimize contamination from room air.
Before picking up a pipette or Pipet-Aid, use two hands to loosen the screw tops of any containers you will be using.
For conical tubes, unscrew the cap with thumb and index finger while holding the tube in your hand. Lift the cap from the tube in thumb and index finger while holding the tube in the same hand. With practice, you will be able to remove the top without dropping the tube or touching the tube rim. Alternatively, the tube may be kept in a rack while removing and replacing the top.
For larger bottles, unscrew the top, then grasp the top between index and middle fingers and lift it off. This leaves the thumb and index fingers free to grab the sides of the bottle. The bottle may then be lifted or tilted without setting the top down.
Note: An inverted top collects particulate contaminants. Keep tops upright (facing down) at all times. If a top must be temporarily put down, place it face down on a disinfected area of the hood's work surface.
Close the bottle tightly before removing from the culture hood,
Before using bottles for media, rinse glassware and bottle tops 5 times in distilled water followed by 5 times in deionized water (ddH2O). Let stand while cleaning additional glassware then pour out any residual water.
Note: Rinse used media bottles in distilled water soon after emptying them of media to prevent the buildup of dry contaminants.
Thread the top onto the bottle so that it is attached but loose enough for steam to escape. Cover the cap and bottle top with aluminum foil. Insure that the foil overlaps at least the top 1/4 of the bottle, is free of tears and holes, and is loose enough for steam to escape beneath the foil.
Place a short strip of autoclave tape across the top of the foil and autoclave on a dry/gravity cycle.
After cooling, gently tighten the bottle top through the foil and store with foil cover in a closed cabinet or on a shelf in a low-traffic area.
Note: Once sterilized, bottles should only be opened and closed in the culture hood.
Fill a small glass or autoclave-compatible plastic beaker with microcentrifuge tubes and cover with a double layer of aluminum foil insuring adequate overlap of the beaker rim.
Affix a strip of autoclave tape and autoclave on dry/gravity. Store with foil cover in a closed cabinet or on a shelf in a low-traffic area.
Before use, decontaminate the bottom and sides of the beaker with 70% ethanol and place in the culture hood.
To withdraw a tube, gently remove the foil without flattening it and place the tented foil top face down on a disinfected area of the work surface.
Gently shake the container until one tube can be reached without touching any others.
Recover the container with foil and keep it in the culture hood until empty.
Note: Only open the container under the culture hood. If contaminated, or if the foil-cover tears, retrieve a fresh beaker of tubes. Clean, unused tubes may be autoclaved again to sterilize.
Note: Convenient, disposable bottle-top filters are available from a number of manufacturers. Reusable bottle-top filters provide a more cost effective solution for laboratories on a more restricted budget.
|
Figure 17. Add a 0.22 membrane filter to the bottle-top filter housing and loosely assemble the filter housing. Wrap both the filter and a bottle cap in foil and affix with autoclave tape.
Wash and rinse filter holders 5 times in distilled water and 5 times in deionized water as with glass bottles.
Remove the GL45 cap from a glass bottle and screw the bottom of the filter to the bottle.
Use blunt forceps or gloved hands to place a 47 mm diameter, 0.22 µm filter into the holder. Discard any blue filter spacer.
Assemble the filter holder loosely to allow steam to penetrate.
Wrap a piece of aluminum foil vertically around the filter holder and at least the top 1/4 of the bottle. Pinch and roll the two ends of the foil together. Cover the top of the filter with another piece of foil. Hand-crimp the foil to the bottle at the bottom. Tape the foil to the bottle with short strips of autoclave tape.
Note: To prevent foil tearing, align the folded seam with the filter's vacuum connector.
Place a GL45 bottle cap you removed earlier face down on a double-layered piece of foil. Wrap the foil around the cap completely and affix with autoclave tape.
Autoclave the filter and cap on a dry/gravity cycle and store with the foil covers in a closed cabinet or on a shelf in a low-traffic area.
Before use, unwrap the filter holder in the flow hood under sterile conditions and tighten all fittings by hand.
Attach the vacuum line and filter a small quantity of media into the filter to test for leaks. If no leaks were found, pour the remaining media to be filtered into the reservoir under vacuum.
After filtering media, remove the filter, unwrap the bottle cap under the culture hood, and close the bottle.
Note: If the filter becomes clogged, remove the vacuum line and gently unscrew the filter from the sterile bottle. Pour the remaining liquid from the filter reservoir back into the source bottle. Temporarily cap the already filtered liquid. Use a fresh bottle-top filter to process the remainder of the liquid and sterilely pour the two filtered contents together.
Place a wrapped syringe filter and appropriately sized syringe in the culture hood.
Syringe filters are wrapped with molded polystyrene covering the outlet side and a flat paper/foil cover on the inlet side. Remove the paper/foil cover but do not remove the filter from the polystyrene wrapping.
Unwrap the syringe and pull the plunger completely out. The plunger may be temporarily placed back within the syringe wrapping or upside-down on the disinfected work surface.
Twist the syringe tip onto the filter top until snug.
While holding the syringe, lift the filter out of its plastic wrapping. Do not touch the filter disc.
Pour the liquid to be filtered into the upright syringe.
Gently replace the plunger onto the top of the syringe while holding the assembly over the destination container.
Gently press the plunger until a seal is made, then increase pressure to filter the remaining liquid.
Cap the container and discard the filter and syringe according to according to your institution's safety policies.
Note: Syringe filters always retain some residual unfiltered liquid. To avoid contamination, do not attempt to force this remaining liquid through the filter.
Only open serological pipettes under the culture hood. Insure that there are no tears or cuts in the pipette wrapping. If the Pipet-Aid is stored outside of the hood, disinfect with 70% ethanol and wipe before placing it in the culture hood.
With one hand, grasp the top of the wrapping. In the other hand, grasp the pipette upright with thumb pressing vertically against the clear plastic side.
While placing pressure on the pipette with the thumb, use the other hand to pull the top of the wrapping toward the thumb. This punctures the top of the pipette through the paper side of the wrapping. Fold the wrapping down without touching the pipette.
Insert the pipette into the Pipet-Aid and twist to seat. Turn the pipette until the markings are easily readable and remove from the wrapping.
Dip only the tip of the pipette into the source liquid without touching the rim of the container.
Aspirate the desired amount. Watch the liquid level in the pipette to prevent overfilling and wetting the cotton plug and Pipet-Aid.
Note: Gloves, pipettes, bottles, etc. pick up particulate matter while outside of the hood and shed it inside of the hood. Do not pass hands, bottles, etc. over the mouth of any open container. When pipetting, tilt both the container and the pipette at an angle so that neither the Pipet-Aid nor the hands are directly above the container mouth.
Dispense the contents above any existing liquid without touching the rim of the destination container or any existing contents inside of the destination container.
Note: For accurate micropipetting, dispense the liquid with the pipette tip touching the inside rim of the destination container. Discard the tip.
To minimize back-contamination, dip the pipette back into the source container only if you are sure that it did not touch any other liquid or surface during the transfer.
When changing liquids or if the pipette becomes contaminated, discard the pipette and use a fresh pipette. Use the same care with pipette tips.
Note: Watch the pipette at all times before and during liquid transfer to insure that it does not touch the outside of any container or any surface.
Discard of used pipettes according to your institution's safety policies.
Hold the source bottle far enough above the destination bottle so that the rims of the two never touch.
Gently pour the source liquid into the destination container with one smooth motion.
Note: Wipe up any spills immediately. Blot the spill dry. Decontaminate the spill area with 70% ethanol and wipe dry with Kimwipes.
Fill stainless steel boxes or cylinders with Pasteur pipettes, narrow end down.
Tape the lid to the side of the box with a short strip of autoclave tape and autoclave on dry/gravity.
Keep at least one autoclaved box in the culture hood. Cut or remove the autoclave tape before use.
When ready to aspirate, turn on the vacuum supply. If the vacuum valve or switch is outside of the culture hood, decontaminate gloves with ethanol or change them.
Grasp the Pasteur pipette box horizontally with both hands and remove the top.
Shake the pipette box horizontally until one pipette is extended more than any others.
Grasp the end of this pipette between two free fingertips and withdraw it.
Note: Alternately, with the hand facing away from the box, grasp the end of the pipette between the backs of two fingers, and withdraw the pipette. This orientation prevents the pipette tip from touching any contaminating surface while the box is reassembled.
Carefully place the lid back on the box and put the box down.
While touching only the top end of the pipette, insert it into the Tygon tubing on the vacuum trap.
Aspirate the liquid from the top down, touching only the inside of the container with the pipette tip.
Change pipettes between samples to avoid cross contamination of experiments.
Discard of used glass pipettes according to your institution's safety policies.
Adams, M. C., Matov, A., Yarar, D., Gupton, S. L., Danuser, G., and Waterman-Storer, C. M. (2004). Signal analysis of total internal reflection fluorescent speckle microscopy (TIR-FSM) and wide-field epi-fluorescence FSM of the actin cytoskeleton and focal adhesions in living cells. J Microsc 216, 138-152. Abstract Article
Axelrod, D. (2001). Total internal reflection fluorescence microscopy in cell biology. Traffic 2, 764-774. Abstract Article
Barker, K. (2004). At The Bench: A Laboratory Navigator, Updated Edition (New York: Cold Spring Harbor Laboratory Press).
Bianchi, L., and Driscoll, M. (2006). Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.122.1, http://www.wormbook.org. Article
Bloom, L. (1993). Genetic and molecular analysis of genes required for axon outgrowth in Caenorhabditis elegans. PhD dissertation. Massachusetts Intitute of Technology, Cambridge MA. http://hdl.handle.net/1721.1/12667
Burkel, B. M., von Dassow, G., and Bement, W. M. (2007). Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 64, 822-832. Abstract Article
Casbas-Hernandez, P., Fleming, J. M., and Troester, M. A. (2011). Gene expression analysis of in vitro cocultures to study interactions between breast epithelium and stroma. J. Biomed. Biotechnol. 2011, 520987. Abstract Article
Choe, K. P., and Strange, K. (2008). Genome-wide RNAi screen and in vivo protein aggregation reporters identify degradation of damaged proteins as an essential hypertonic stress response. Am. J. Physiol., Cell Physiol. 295, C1488-1498. Abstract Article
Christensen, M., Estevez, A., Yin, X., Fox, R., Morrison, R., McDonnell, M., Gleason, C., Miller, D. M., and Strange, K. (2002). A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33, 503-514. Abstract Article
Christensen, M., and Strange, K. (2001). Developmental regulation of a novel outwardly rectifying mechanosensitive anion channel in Caenorhabditis elegans. J. Biol. Chem. 276, 45024-45030. Abstract Article
Cowan, A. E., and McIntosh, J. R. (1985). Mapping the distribution of differentiation potential for intestine, muscle, and hypodermis during early development in Caenorhabditis elegans. Cell 41, 923-932. Abstract Article
Cox, G. N., Staprans, S., and Edgar, R. S. (1981). The cuticle of Caenorhabditis elegans. II. Stage-specific changes in ultrastructure and protein composition during postembryonic development. Dev. Biol. 86, 456-470. Abstract Article
Danuser, G., and Waterman-Storer, C. M. (2006). Quantitative fluorescent speckle microscopy of cytoskeleton dynamics. Annu. Rev. Biophys. Biomol. Struct. 35, 361-387. Abstract Article
Edgar, L. G., and Goldstein, B. (2012). Culture and manipulation of embryonic cells. Methods Cell Biol. 107, 151-175. Abstract Article
Edgar, L. G., and McGhee, J. D. (1988). DNA synthesis and the control of embryonic gene expression in C. elegans. Cell 53, 589-599. Abstract Article
Fisher, J., Piterman, N., Hubbard, E. J., Stern, M. J., and Harel, D. (2005). Computational insights into Caenorhabditis elegans vulval development. Proc. Natl. Acad. Sci. U. S. A. 102, 1951-1956. Abstract Article
Fox, R. M., Von Stetina, S. E., Barlow, S. J., Shaffer, C., Olszewski, K. L., Moore, J. H., Dupuy, D., Vidal, M., and Miller, D. M. (2005). A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics 6, 42. Abstract Article
Goldstein, B. (1992). Induction of gut in Caenorhabditis elegans embryos. Nature 357, 255-257. Abstract Article
Goodman, M. B., Hall, D. H., Avery, L., and Lockery, S. R. (1998). Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron 20, 763-772. Abstract Article
Hajduskova, M., Ahier, A., Daniele, T., and Jarriault, S. (2012). Cell plasticity in Caenorhabditis elegans: from induced to natural cell reprogramming. Genesis 50, 1-17. Abstract Article
Harrison, R. G., Greenman, M., Mall, F. P., and Jackson, C. (1907). Observations of the living developing nerve fiber. The Anatomical Record 1, 116-128. Article
Hu, K., Ji, L., Applegate, K. T., Danuser, G., and Waterman-Storer, C. M. (2007). Differential transmission of actin motion within focal adhesions. Science 315, 111-115. Abstract Article
Husson, S. J., Landuyt, B., Nys, T., Baggerman, G., Boonen, K., Clynen, E., Lindemans, M., Janssen, T., and Schoofs, L. (2009). Comparative peptidomics of Caenorhabditis elegans versus C. briggsae by LC-MALDI-TOF MS. Peptides 30, 449-457. Abstract Article
Hyenne, V., Tremblay-Boudreault, T., Velmurugan, R., Grant, B. D., Loerke, D., and Labbe, J. C. (2012). RAB-5 controls the cortical organization and dynamics of PAR proteins to maintain C. elegans early embryonic polarity. PLoS ONE 7, e35286. Abstract Article
Johnston, W. L., and Dennis, J. W. (2012). The eggshell in the C. elegans oocyte-to-embryo transition. Genesis 50, 333-349. Abstract Article
Kaji, H., Camci-Unal, G., Langer, R., and Khademhosseini, A. (2011). Engineering systems for the generation of patterned co-cultures for controlling cell-cell interactions. Biochim. Biophys. Acta 1810, 239-250. Abstract Article
Khan, F. R., and Mcfadden, B. A. (1980). A rapid method of synchronizing developmental stages of Caenorhabditis elegans. Nematologica 26, 280-282. http://dx.doi.org/10.1163/187529280X00189 Article
Killeen, M. T., and Sybingco, S. S. (2008). Netrin, Slit and Wnt receptors allow axons to choose the axis of migration. Dev. Biol. 323, 143-151. Abstract Article
Klass, M. R., and Hirsh, D. (1981). Sperm isolation and biochemical analysis of the major sperm protein from Caenorhabditis elegans. Dev. Biol 84, 299-312. Abstract Article
Kline, D. (2009). Quantitative microinjection of mouse oocytes and eggs. Methods Mol. Biol. 518, 135-156. Abstract Article
Lamitina, S. T., Morrison, R., Moeckel, G. W., and Strange, K. (2004). Adaptation of the nematode Caenorhabditis elegans to extreme osmotic stress. Am. J. Physiol., Cell Physiol. 286, C785-791. Abstract Article
Laufer, J., Bazzicalupo, P., and Wood, W. (1980). Segregation of developmental potential in early embryos of Caenorhabditis elegans. Cell 19, 569-577. Abstract Article
Laufer, J. S. (1982) Cell Determination in early embryos of the nematode Caenorhabditis elegans. PhD dissertation, University of Colorado, Boulder CO.
Lin, S., and Talbot, P. (2011). Methods for culturing mouse and human embryonic stem cells. Methods Mol. Biol. 690, 31-56. Abstract Article
Lin, X.-G., Ming, M., Chen, M.-R., Niu, W.-P., Zhang, Y.-D., Liu, B., Jiu, Y.-M., Yu, J.-W., Xu, T., and Wu, Z.-X. (2010). UNC-31/CAPS docks and primes dense core vesicles in C. elegans neurons. Biochem. Biophys. Res.Commun. 397, 526-531. Abstract Article
Moldaver, M. V., and Yegorov, Y. E. (2009). Sparse plating increases the heterogeneity of proliferative potential of fibroblasts. Mech. Ageing Dev. 130, 337-342. Abstract Article
Nass, R., and Hamza, I. (2007). The nematode C. elegans as an animal model to explore toxicology in vivo: solid and axenic growth culture conditions and compound exposure parameters. Curr. Protoc. Toxicol. Chapter 1, Unit1.9. Abstract Article
Nemethova, M., Auinger, S., and Small, J. V. (2008). Building the actin cytoskeleton: filopodia contribute to the construction of contractile bundles in the lamella. J. Cell Biol. 180, 1233-1244. Abstract Article
Pasapera, A. M., Schneider, I. C., Rericha, E., Schlaepfer, D. D., and Waterman, C. M. (2010). Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877-890. Abstract Article
Portela, V. M., Zamberlam, G., and Price, C. A. (2010). Cell plating density alters the ratio of estrogenic to progestagenic enzyme gene expression in cultured granulosa cells. Fertil. Steril. 93, 2050-2055. Abstract Article
Quinn, C. C., and Wadsworth, W. G. (2008). Axon guidance: asymmetric signaling orients polarized outgrowth. Trends Cell Biol. 18, 597-603. Abstract Article
Richmond, J. E., and Jorgensen, E. M. (1999). One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat. Neurosci. 2, 791-797. Abstract Article
Rohrbach, A. (2000). Observing secretory granules with a multiangle evanescent wave microscope. Biophys. J. 78, 2641-2654. Abstract Article
Schlaeger, E. J. (1996). Medium design for insect cell culture. Cytotechnology 20, 57-70. Abstract Article
Shim, Y.-H., and Paik, Y.-K. (2010). Caenorhabditis elegans proteomics comes of age. Proteomics 10, 846-857. Abstract Article
Spencer, W. C., Zeller, G., Watson, J. D., Henz, S. R., Watkins, K. L., McWhirter, R. D., Petersen, S., Sreedharan, V. T., Widmer, C., Jo, J., et al. (2011). A spatial and temporal map of C. elegans gene expression. Genome Res. 21, 325-341. Abstract Article
Sternberg, P. W. (2005). Vulval development. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.6.1, http://www.wormbook.org. Abstract Article
Strange, K., Christensen, M., and Morrison, R. (2007). Primary culture of Caenorhabditis elegans developing embryo cells for electrophysiological, cell biological and molecular studies. Nat. Protoc. 2, 1003-1012. Abstract Article
Sulston, J. E., and Horvitz, H. R. (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110-156. Abstract Article
Sund, S. E., Swanson, J. A., and Axelrod, D. (1999). Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys. J. 77, 2266-2283. Abstract Article
Szilagyi, M. Gehman, E., LaPenotiere, H., Lewis, J., Clegg, E., and Jackson, D.A. (2006). Global alterations in gene expression during organophosphate pesticide intoxication and recovery: Interim report. USAMRICD-TR-06-02 (U.S. Army Medical Research Institute of Chemical Defense). http://www.dtic.mil/cgi-bin/GetTRDoc?AD=ADA469210
Varshney, L. R., Chen, B. L., Paniagua, E., Hall, D. H., and Chklovskii, D. B. (2011). Structural properties of the Caenorhabditis elegans neuronal network. PLoS Comput. Biol. 7, e1001066. Abstract Article
Von Stetina, S. E., Watson, J. D., Fox, R. M., Olszewski, K. L., Spencer, W. C., Roy, P. J., and Miller, D. M. (2007). Cell-specific microarray profiling experiments reveal a comprehensive picture of gene expression in the C. elegans nervous system. Genome Biol. 8, R135. Abstract Article
Waters, K. A., and Reinke, V. (2011). Extrinsic and intrinsic control of germ cell proliferation in Caenorhabditis elegans. Mol. Reprod. Dev. 78, 151-160. Abstract Article
White, J. G., Southgate, E., Thomson, J. N., and Brenner, S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 314, 1-340. Abstract Article
Wilson, H. (1907). On some phenomena of coalescence and regeneration in sponges. J. Exp. Zool. 5, 245-258. Article
*Edited by Oliver Hobert. Last revised November 14, 2012. Published February 21, 2013. This chapter should be cited as: Zhang S. and Kuhn JR. Cell isolation and culture (February 21, 2013), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.157.1, http://www.wormbook.org.
Copyright: © 2012 Zhang S. and Kuhn JR. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: jrkuhn@vt.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.