Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard Schierenberg
Table of Contents
Two methods are available for the generation of transgenic C. elegans: microinjection and microparticle bombardment (see Transformation and microinjection). Until quite recently, the only one-step method for producing integrated transgenes, as opposed to extrachromasomal arrays, has been the microparticle bombardment method. This method has long been used to transform plant tissues and mammalian tissue slices, but was first published for use in C. elegans in 2001 (Lo et al., 1994; Lorence and Verpoorte, 2004; Praitis et al., 2001). Since its introduction the bombardment technique has grown in popularity and is widely used to generate low copy-number transgenes integrated randomly within the C. elegans genome. Often called biolistic transformation, the technique involves accelerating micron sized gold particles coated with DNA (bullets) toward a living target, essentially a “gene gun”. Most modern biolistic devices (e.g., Biorad PDS-1000/He Hepta) use high-pressure helium gas to accelerate the particles within a vacuum chamber, targeting the biological specimens to be transformed, in this case a large population of adult C. elegans hermaphrodites (Fitzpatrick-McElligott, 1992; Praitis, 2006). The gold particles become lodged randomly within the cells of the target animals. After several generations, heritably transformed animals can be recovered, presumably due to the small fraction of DNA coated gold particles that lodge within the germ cells of the parent animals.
This technique allows for the recovery of both low copy-number, randomly integrated, transgenes, as well as high copy-number extrachromasomal arrays (Praitis, 2006; Praitis et al., 2001). The bombardment technique is not efficient on a per-animal basis and therefore depends upon genetic selection, usually accomplished by including a wild-type copy of the unc-119 gene with the DNA of interest, and targeting a population of unc-119 mutant animals. unc-119 mutants grow slowly, are mostly paralyzed, and fail to form dauer larvae under starvation conditions. Selection for movement and formation of dauers after starvation is very efficient and allows for facile recovery of transformed animals. The C. briggsae version of the unc-119 gene (2.2 kb) is smaller than the C. elegans version (5.5 kb) due to smaller introns, but rescues the C. elegans unc-119 mutant extremely well (Maduro and Pilgrim, 1996). Therefore the C. briggsae version of the unc-119 gene, rather than the C. elegans unc-119 gene, is found in some C. elegans expression vectors. Recent publications also report successful recovery of transgenic C. elegans lines after bombardment using drug resistance markers (Semple et al., 2012).
Low copy-number transgenes obtained by microparticle bombardment offer several advantages that make them experimentally useful. The first advantage is that at least some of the low-copy lines obtained by this method allow stable expression of the transgenes in the C. elegans germline (Transgenic solutions for the germline; Praitis, 2006; Praitis et al., 2001). Transgenes are prone to silencing in C. elegans germ cells. In fact simple extrachromasomal arrays are almost universally silent in the germ line (Kelly et al., 1997). For somatic cell expression, the low-copy transgenes produced by bombardment generally offer relatively even expression among equivalent animals in a population and among equivalent cells within a single animal (Praitis et al., 2001). Other methods that can also improve germline expression of transgenes and the reproducibility of somatic cell expression include complex extrachromosomal arrays, and single copy integrants (Mosci method) (Frokjaer-Jensen et al., 2012; Frokjaer-Jensen et al., 2008; Kelly et al., 1997; Zeiser et al., 2011).
• NGM (Maintenance of C. elegans) or NGM-lite (Eric Lambie, personal communication; Notes on worm plates; MYOB and a happier medium) C. elegans growth plates in 6 cm, 10 cm, and 15 cm sizes
• M9 salts (sterile)
• 2.5 M CaCl2 (sterile)
• 2xYT bacterial growth media (sterile)
• 10 mg/ml nystatin (Calbiochem 475914)
• 100 mg/ml streptomycin (Sigma-Aldrich® S6501)
• 0.1 M spermidine in 200 μl aliquots (Sigma-Aldrich S-0266)
• 6 ml egg media aliquots
• 70% ethanol
• 100% ethanol
• 60 mg/ml Gold Particles (Biorad 165-2263 1.0 μm, or DMC custom 2.0 μm made by dmc2 Metals Group, South Plainfield, NJ 07080, 908-561-1100) in 50% glycerol
• Hepta stopping screens - autoclaved (Biorad 165-2226)
• 1350 psi rupture disks (Biorad 165-2330)
• Macrocarriers (Biorad 165-2335)
• L-shaped Plastic Spreaders, Sterile (VWR 30002-120)
• 1.7 ml SlickSeal™ Tubes (VWR 20172-945)
• disposable 5 ml and 10 ml pipets
• HB101 E. coli (CGC)
• Biolistic PDS-1000/He particle delivery system with Hepta Adapter (Biorad 165-2258)
• Savant VLP 200 Vacuum Pump (or equivalent)
• Waring® Blender (e.g., PBB2 Professional Bar Blender from Amazon)
• Vortex-Genie 2 with Foam Tube Holder Insert
• Pipet-Aid®
• forceps
Here we describe a version of the microparticle bombardment protocol that we have developed over the last 10 years, derived in large part by combining aspects of existing protocols obtained from the Geraldine Seydoux, Shai Shaham, and Ronald Plasterk labs. This variation of the bombardment protocol has been employed in our laboratory to deliver hundreds of different DNA constructs into C. elegans and generally produces 10-20 independent transformed lines per bombardment. Roughly half of these lines are integrated, and more than half of the integrated lines are typically viable as homozygotes. We generally perform 3-4 bombardments on one day of each week, although this throughput depends upon continual passaging and cleaning of unc-119 mutant stocks. Roughly 3 ml of packed adult hermaphrodites are required for one successful bombardment, so large scale growth of unc-119 mutants is the most time-consuming and laborious step in the protocol.
Autoclave the metal carafe of a Waring blender, or sterilize a glass blender carafe by running with 10% bleach, followed by rinsing with sterile water. Spray the outer shell of raw chicken eggs obtained from the grocery store with 70% ethanol, and wipe away the liquid using Kimwipes™. Crack the eggs and collect their contents in a sterile beaker. Discard the shells. Boil 25 ml of water per egg in an Erlenmeyer flask in a microwave oven. Pour the contents of the eggs into the blender, add the boiling water, and run the blender. When the solution becomes homogenous, dump the contents of the blender into a sterile beaker, and let the froth settle for one hour. Pipette the egg solution into 6 ml aliquots in 15 ml conical tubes, and incubate the tubes at 65°C for 1 hour in a water bath to inactive endogenous lysozyme. Allow the tubes to cool to room temperature, and freeze them for long-term storage at −80°C.
A concentrated HB101 E. coli stock for egg plates is prepared by seeding 250 ml of 2xYT medium, in a 2 L flask, with a single HB101 colony. Shake the flask overnight (37°C at 225 rpm). The following day, aliquot the culture into 50 ml conical tubes. Before use, pellet the bacteria at 4000 rpm for 15 minutes. Discard the supernatant. Resuspend the bacterial pellet by vortex in a volume of M9 buffer roughly equal to the volume of the pellet. Store the unused aliquots of bacteria at 4°C. HB101 cultures used for seeding smaller growth plates, prior to the egg plate stage, are not concentrated.
Prepare starter cultures of DP38 unc-119(ed3) worms on 6 cm NGM plates that have been seeded with an HB101 lawn. HB101 is preferred in this protocol over the typical OP50 strain of E. coli used for C. elegans genetics, because HB101 produces a thicker lawn (more food for the animals), and because HB101 is an easier strain for the mutant animals to ingest (see C. elegans feeding). Some protocols for large scale growth of C. elegans call for the use of growth plates highly enriched in peptone, but we have found that unc-119 mutants grow best on standard NGM or NGM-lite (Maintenance of C. elegans). unc-119 mutants also grow better at low temperature (15-20°C) than high temperature (25°C). Allow the worms to starve out the plates. Divide one complete, recently starved, 6 cm plate to four new 10 cm HB101 seeded NGM/NGM-lite plates. Spread small chunks containing unc-119 worms to several locations on the large HB101 lawn to facilitate even distribution of the worms as they grow, since unc-119 worms are nearly paralyzed and never stray very far from the area in which they hatch. For the same reason take care to place the chunks face down, with starved worms in direct contact with the bacterial lawn. Allow the 10 cm plates to exhaust their food supply, typically nine to eleven days at 20°C. Starved worm cultures can be saved at 15-20°C for up to 2 weeks prior to their use in inoculating egg plate cultures. Discard any plates bearing visible contamination before starting egg-plate cultures.
Note: The growth of starter cultures of unc-119 mutant worms is quite time consuming, generally taking 2-3 weeks for completion. If bombardments are to be performed fairly regularly, then 6 cm and 10 cm NGM/HB101 cultures of unc-119 mutant worms can be started by chunking on a weekly basis. This approach requires a relatively small effort, and creates a situation in which there are always worms available to use to start egg plate cultures, reducing the required lead-time for bombardment to 4 days. Because of their paralysis and slow growth, unc-119 mutant cultures are more prone than wild-type cultures to accumulate contaminating mold and bacteria, and must be cleaned by basic hypochlorite digestion when contamination occurs (Maintenance of C. elegans).
Four 15 cm egg plate cultures are required to obtain enough animals for one bombardment. One 10 cm culture is used to seed one 15 cm egg plate culture. For one bombardment, wash worms from four recently starved 10 cm unc-119 worm cultures. Wash the plates twice, using 5 ml of 100 μg/ml streptomycin-spiked M9 salts per 10 cm plate, collecting the liquid and worms in one 50 ml conical tube. Next, pellet the worms by centrifugation for 2 min. at 1500 rpm. Discard the supernatant.
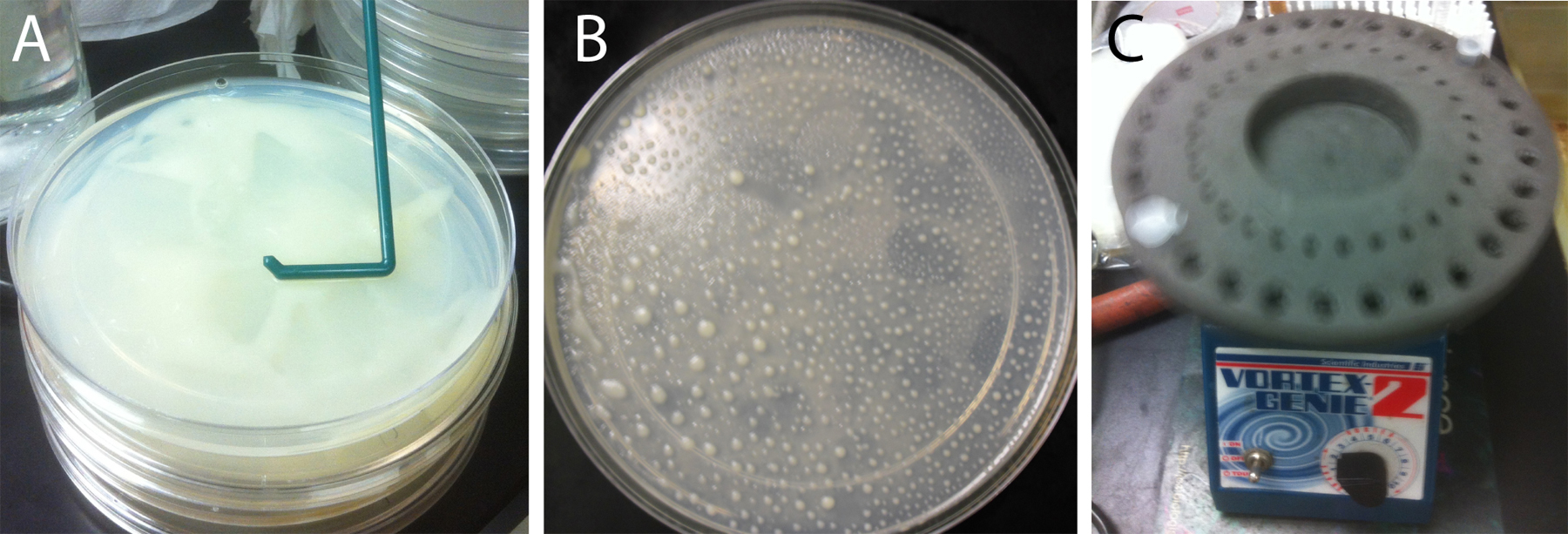
Spin down one 50 ml tube of HB101 bacteria per set of four egg plates at 3000 rpm for 5 min. Discard the supernatant. Add 500 μl of the streptomycin-spiked M9 buffer to the pellet and resuspend the bacteria by brief vortexing. Mix the resuspended bacteria with one thawed 6 ml aliquot of egg medium, then mix it with the worm pellet obtained from four starved 10 cm culture plates. Aliquot 25% of the worm/HB101/egg mixture onto the surface of each of four 15 cm NGM lite plates. Use a disposable bacterial spreader to evenly distribute the worm/HB101/egg mixture on the plate (Figure 1A). It is necessary to mix the worms with the bacterial media prior to spreading because the nearly paralyzed unc-119 strain cannot evenly self-distribute. Grow the unc-119 egg plate cultures for approximately 4 days at 20°C, until most animals are gravid adults, before using the worms for bombardment. The time to harvest can vary and must be determined by inspection of the cultures (see Figure 1B).
|
Figure 1. Growth of unc-119(ed3) worms for bombardment. (A) Spreading mixture of worms/egg/HB101 across surface of 15 cm NGM-lite plate. (B) Plate ready to harvest for bombardment. After spreading, grown for 4 days at 20°C. Note that worms are found mostly in large piles with remaining food. The majority of the animals are gravid adults. (C) Vortexing gold particles at low speed using foam tube holder insert. A vortexer with such an insert is required equipment for the long vortexing steps associated with precipitation of DNA onto the gold particles.
Large scale unc-119 worm cultures can be grown in liquid (S medium, see Maintenance of C. elegans), or on a very large number of more standard 10 cm HB101 seeded growth plates (without egg media, see Transgenic solutions for the germline). Liquid cultures typically require a shaking incubator equipped with refrigeration to maintain growth temperatures ≤ 20°C. In our experience the egg plate method requires the least effort to produce the large number of animals required for successful bombardment.
Use a siliconized microcentrifuge tube to prepare gold particles. Put the siliconized microcentrifuge tube on a balance, and zero the balance. Weigh out 30 mg of gold particles in the tube. Add 1 ml of 70% ethanol. Vortex the tube for 5 min., using a foam insert in the vortexer to hold the tubes (Figure 1C). Stop the vortexer and allow the tube to rest for 15 min. Spin the tube briefly and discard the supernatant. Wash the particles three times with 1 ml of sterile water by adding the water, vortexing, and then tap spinning the tube (i.e., spin very briefly using the “pulse” button on the centrifuge). When removing the wash water, touch the pipette tip to the side of the tube opposite the gold. After removing the water from the third wash, resuspend the gold in 0.5 ml 50% sterile glycerol. The final gold concentration is 60 mg/ml. The gold may be stored for at least 1-2 months at 4°C. 50% glycerol stocks should be stored in the dark.
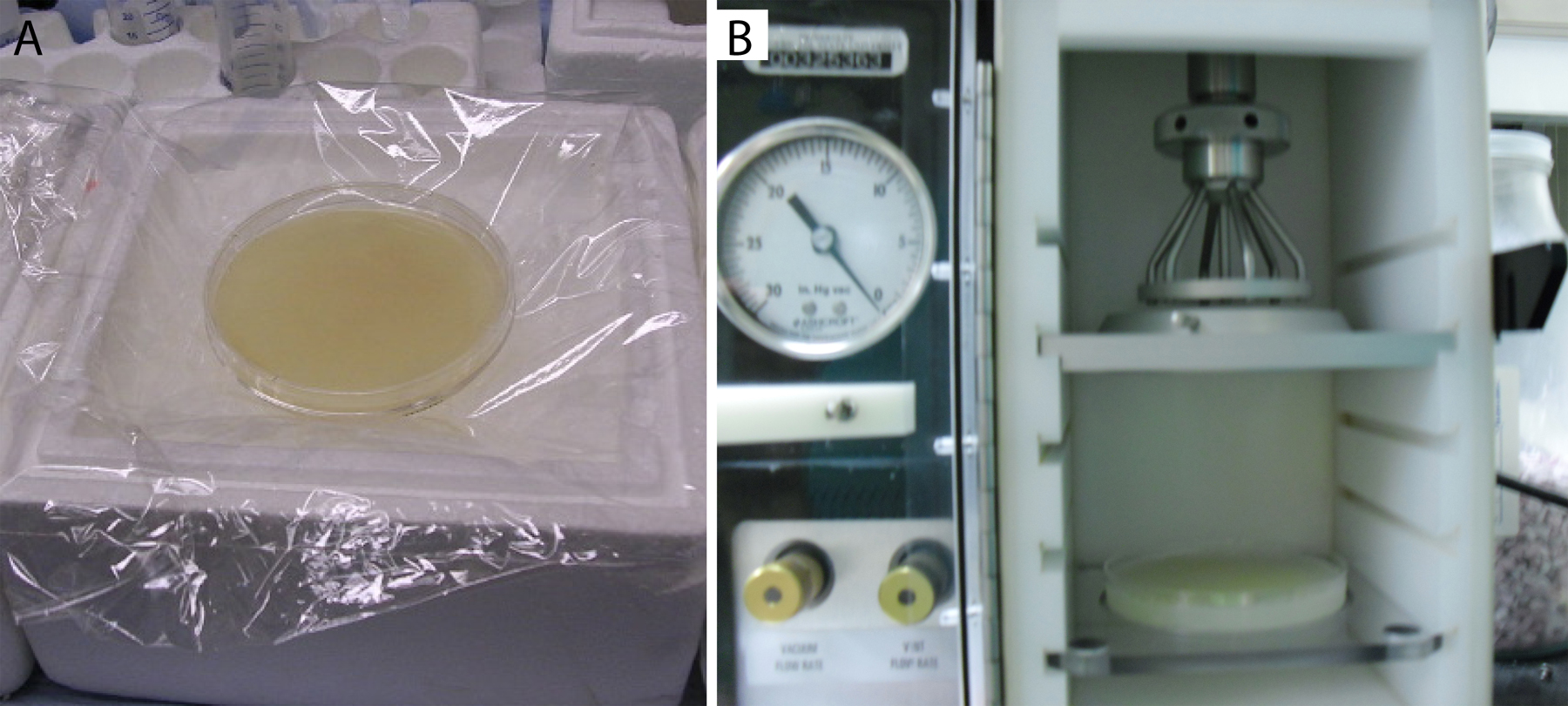
Worms grown on four egg plates are harvested in buffer and placed on one seeded 10 cm NGM/NGM-lite plate. This process requires that the receiving NGM plate be dry enough to soak up the transferred buffer, such that one ends up with very tight packing of the worms to be bombarded. In order to ensure that the 10 cm NGM plates to be bombarded will not be too wet, they must be dried prior to application of worms in a 37°C incubator, with the lid slightly ajar. Drying generally takes 2-4 hours. Afterwards, place the plates on Saran™ wrap covered ice to chill (Figure 2A). Chilling the plates keeps the worms from moving prior to bombardment, keeping them in a tightly packed monolayer, or even bilayer, covering the entire plate surface. The Saran wrap helps prevent contamination of the worms by the ice, which tends to be a fruitful source of unwanted bacteria and mold (Figure 2A).
|
Figure 2. Worms ready to bombard. (A) Approximately 3 ml of packed worms coating the surface of a chilled 10 cm NGM-lite plate (HB101 seeded). A confluent layer of worms completely coats the surface of the plate. Chilling the plate stops worm movement that creates gaps in the coverage. Plastic wrap covering the ice reduces contamination of the worms with bacterial and fungal microbes. (B) Confluent worm plate in bombardment chamber. The plate is attached to the shelf using at least five pieces of rolled up lab tape. Note position relative to pressure divider and macrocarrier holder components of Hepta adapter.
Check stock of 2.5 M CaCl2 for undissolved precipitate. If the solution contains undissolved precipitate, warm it in a 37°C water bath until the precipitate re-enters solution. Thaw a 0.1 M spermidine aliquot from −80°C stock. Mix 8-16 μg of plasmid DNA, in a total volume of 50 μl, with 100 μl of well-suspended gold particle solution in 50% glycerol, using a siliconized microfuge tube. If an unc-119(+) gene is already present in the plasmid to be used in bombardment, and the total size of the plasmid is less than 10kb, then 10 μg of DNA is a good amount to use. For larger plasmids, a proportionately greater amount of DNA is often required to obtain good results. Also, if the unc-119(+) gene is on a separate plasmid from the one you wish to bombard, using a mixture of two plasmids (8-10 μg of each) is best. Co-bombardment of two plasmids works, but only about two-thirds of the integrated lines contain the unselected plasmid. Integration of a rescuing unc-119 gene within the plasmid of interest improves the efficiency of useful transgene recovery.
After combining the gold particle solution and DNA, vortex the tube for at least 1 minute (Figure 1C). Next, add 150 μl 2.5M CaCl2. Vortex the solution again, for at least 1 minute. Then, add 60 μl 0.1M spermidine. Vortex the tube 3 to 5 min. (Figure 1C). Re-freeze remaining spermidine stock (store frozen at −80°C). Tap spin the solution. Pulse to 6,000 rpm then release. Spinning the solution too long or at much higher g-force will make resuspension difficult or impossible. After the tap spin, remove the supernatant. Add 300 μl of 70% ethanol. Vortex the solution very well. The precipitated DNA makes the gold very sticky. Tap spin the solution again. Remove the supernatant. Then, add 170 μl of 100% ethanol, and vortex the tube on high for 5 to 10 min. Then, turn down the vortex to a low speed until the DNA-coated gold is actually used (Figure 1C). All vortexing steps are typically performed using a foam insert in the vortexer that can hold the tubes without supervision (Figure 1C). We find that a rubber mouse pad placed under the vortexer can prevent its migration across the bench during the long mixing steps.
Note: The DNA precipitation protocol is based upon a DNA starting volume of 50 μl. If using a larger starting volume increase CaCl2, spermidine, and ethanol (but not gold particle) volumes proportionately.
Add some dryerite covered by a Kimwipe to an empty 15 cm petri dish. With forceps, dip seven macrocarriers per bombardment, one at a time, into 100% ethanol and lay them on the kimwipe in the plate to dry (Figure 3A). Keep the lid slightly ajar in order to let the macrocarriers dry.
|
Figure 3. Preparing Macrocarriers. (A) Drying macrocarriers after dipping in ethanol. (B) Pipetting DNA-coated gold particles onto macrocarriers. (C) Drying DNA-coated gold particles on macrocarriers. Note that gold particles are centered over holes in macrocarrier holder. (D) Pressing tool used to firmly seat macrocarriers in macrocarrier holders. Apply pressure while using a twisting motion to firmly seat the macrocarriers such that they remain in place when carrier is inverted.
Clean the bombardment chamber with ethanol and Kimwipes (Figure 4A). Wash the target plate shelf in a similar manner (Figures 2B and 4A). Roll up four or five pieces if lab tape, with the sticky part on the outside, in order to tape the worm plate to the shelf. Place the shelf in the bottom slot of the bombardment chamber (Figure 2B). Wipe down the metal parts of the macrocarrier holders with ethanol and Kimwipes (Figure 3B). Load the macrocarriers into the lid of the macrocarrier holder with forceps. Wipe the red plastic pressing tool with ethanol and Kimwipes (Figures 3D and 4A). Press the macrocarriers into the lid with the red pressing tool using a twisting motion. Add 20 μl of DNA-coated gold particles to the center of each macrocarrier, try to cover only the central area of the macrocarriers which lie above the holes in the metal macrocarrier holder (Figure 3B, C). Then split up any remaining DNA-coated gold particles among the macrocarriers, again only covering the central areas (Figure 3B, C). Let the gold solution on the macrocarriers dry (Figure 3C). A fume hood can speed up the drying process.
Wash unc-119(ed3) mutant worms from 4 egg plates with sterile streptomycin-spiked M9 salts (Figure 1B). It is easiest to cover the worms with M9 buffer and allow them soak for 10 min. or more. Disposable scrapers can be used to dislodge worms. Remove the worms in M9 buffer from the plates with a pipette. Transfer the worms to a 50 ml conical centrifuge tube. Centrifuge the worms for 2 min. at 1500 rpm in a table-top centrifuge. Pipette off the supernatant, and discard. Resuspend the worms in M9 in a total of 25 ml. Allow the adult worms to settle to the bottom of the tube by gravity for 15-30 min. Then, remove the supernatant. The purpose of letting the worms settle by gravity is to enrich for adults over larvae, and to remove egg debris. Pipet the adult worm pellet derived from four 15 cm egg plates onto one dried, ice-chilled, 10 cm HB101 plate (Figure 2A). Draw up the worms from the bottom of the pellet. 2.75 ml of packed worm pellet is the perfect amount for one 10 cm target plate. Let the plate rest on Saran wrap covered ice for at least 30 min. to allow the plates to absorb the liquid. A confluent layer of worms should coat the whole plate, and the worms should not be floating in liquid.
Before an actual bombardment, test firing the apparatus is recommended. To test fire the apparatus, dip a rupture disk in 100% ethanol with forceps and shake off the excess ethanol. Place the rupture disk into the top of the pressure divider (Figure 4B). Screw the pressure divider into the bombardment chamber and tighten using the small torque wrench provided with PDS-1000 (Figure 4C). Close the door of the bombardment chamber. Open the helium and adjust the pressure to at least 200 psi higher than the amount needed to rupture the disk. We usually use a 1350 psi rupture disk, thus requiring at least 1550 psi in pressure from the helium tank. Turn on the vacuum pump. Turn on power to the bombardment chamber (Figure 4A, switch 1). Place the vacuum switch to the vacuum position (Figure 4A, switch 2), and hold it until the needle on the vacuum gauge reaches 28 ”Hg (Figure 4A). Then, very quickly turn the vacuum switch to hold, skipping the vent level of the switch. Press and hold down the fire button, allowing gas pressure to slowly build behind the rupture disk (Figure 4A, switch 3). Gas pressure at the level of the rupture disk is monitored via the helium pressure gauge on top of the PDS-1000 (Figure 4A). Release the fire button once the rupture disk breaks. One should hear a distinct pop. One should also see that the helium pressure gauge on top of the PDS-1000 approached 1350-1500 psi, then suddenly dropped to 0 psi. Turn the vacuum switch to vent, and turn off power to the vacuum pump and PDS-1000 (Figure 4A, D). Unscrew the pressure divider using the torque wrench (Figure 4C). Remove the burst rupture disk using forceps and discard.
Wipe down the two parts of the pressure divider with ethanol and Kimwipes. Dip a rupture disk into ethanol with forceps and shake off the excess ethanol. Place the rupture disk into the pressure divider (Figure 4B). Screw the pressure divider into the nozzle of the bombardment chamber using the torque wrench (Figure 4C). Place an autoclaved stopping screen onto the pegs of the lower half of the macrocarrier holder. Invert the top half of the macrocarrier holder (containing the macrocarriers), and place it onto the pegs of the lower half of the macrocarrier that lies beneath the stopping screen. Place the assembled macrocarrier apparatus onto the metal shelf, and rotate the lever to lock the shelf onto the macrocarrier apparatus. Insert the macrocarrier apparatus shelf complex into the bombardment chamber using the second shelf slot from the top, directly below the Hepta pressure divider (Figure 2B). Rotate the lever of the macrocarrier apparatus such that the holes of the complex align with the holes of the pressure divider. Tape down the worm plate for bombardment to the target plate shelf (Figure 2B). If tape is not used, the plate will jump during rupture of the disk, interfering with successful bombardment.
Close the door of the bombardment chamber. Open the helium, and adjust the pressure to at least 200 psi higher than the amount needed to burst the rupture disk (e.g., 1550 psi). Turn on the vacuum pump (Figure 4D). Turn on power to the bombardment chamber (Fig 4A, switch 1). Place the vacuum switch to the vacuum position (Figure 4A, switch 2), and hold it until the needle on the vacuum gauge reaches 28 ”Hg (Figure 4A). Then, very quickly turn the vacuum switch to hold, skipping the vent level of the switch. Press and hold down the fire button, allowing gas pressure to slowly build behind the rupture disk (Figure 4A, switch 3). Gas pressure at the level of the rupture disk is monitored via the helium pressure gauge on top of the PDS-1000 (Figure 4A). Release the fire button once the rupture disk breaks. One should hear a distinct pop. One should also see that the helium pressure gauge on top of the PDS-1000 approached 1350-1500 psi, then suddenly dropped to 0 psi. Turn the vacuum switch to vent, and turn off power to the vacuum pump and PDS-1000 (Figure 4A, D). Remove the worm plate and set aside at room temperature for at least 30 min. Wash the worms off of the plate with M9/strep, and distribute them equally among twenty 10 cm, HB101-seeded, NGM-lite plates.
While worms are resting, close the helium gas tank and release the remaining pressure in the pressure regulator that is attached to the helium tank. Unscrew the pressure divider using the torque wrench (Figure 4C). Remove the burst rupture disk using forceps and discard. Save the used stopping screen. Used stopping screens may be rinsed and autoclaved for re-use. Wipe down every part of the machine with ethanol and Kimwipes. When finished with all bombardments, jiggle both the vent and fire switches to release any residual helium pressure in the apparatus. This is important. Residual pressure can damage the machine!
Grow the bombarded worms, which are now on twenty-one 10 cm seeded plates (the original plate is saved) at 25°C for 10-14 days or more. Non-Unc transgenic animals can be recovered by simple visual screening, looking for fast moving animals after tapping the plate on the bench or microscope stage. In many cases dozens or hundreds of moving animals are visible. Alternatively recover non-Unc worms by “reverse chunking”, which is performed by placing an agar chunk from a seeded plate, bacterial side up, in the center of the starved bombardment plate. Animals with normal movement will seek out the food on the top of the agar chunk and are easily retrieved the day after placing the bait (Kevin O'Connell, personal communication, see Reverse chunking, a simple and effective method for identifying unc-119 transgenics). Single out non-Uncs from each plate to screen for integrated lines. We generally single out 2 worms per 10 cm plate. We assume that all non-Unc animals from the same founder plate are likely to be siblings carrying the same transgene, although in some cases clear differences in expression level or pattern in animals from the same plate suggest that more than one independent line may be present. If possible we choose larval animals as founders, since adults from such plates are often of a post-reproductive age. Those plates that produce all non-Unc progeny are homozygous integrants. Plates that segregate less than 100% non-Uncs are either heterozygous integrants or extrachromosomal array lines. Heterozygous integrants should segregate about three-fourths non-Uncs and one-fourth Uncs, while arrays vary widely in their maintenance through meiosis with some array lines maintained at around this level. To identify integrated lines initially isolated as heterozygotes, we single out an additional nine non-Unc worms from candidate plates, again assaying in the next generation for 100% transmission of the transgene. We often screen lines for desired expression properties prior to such additional singling out efforts.
We thank Geraldine Seydoux, Shai Shaham, and Ron Plasterk for providing bombardment protocols developed in their laboratories that proved very useful in developing this protocol. We thank Kevin Huang for help with the figures. We also owe thanks to Ken Sato and Miyuki Sato for working out the initial successful version of this protocol in the Grant lab. We thank Richard Davis for suggesting the use of custom made two micron gold particles as DNA carriers for bombardment. This work was supported by NIH grants GM067237 and GM103995 to B.D.G.
Avery, L., and You, Y.J. C. elegans feeding (May 21, 2012), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.150.1, http://www.wormbook.org.
Evans, T.C., ed. Transformation and microinjection (April 6, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.108.1, http://www.wormbook.org.
Fitzpatrick-McElligott, S. (1992). Gene transfer to tumor-infiltrating lymphocytes and other mammalian somatic cells by microprojectile bombardment. Biotechnology 10, 1036-1040. Abstract Article
Frokjaer-Jensen, C., Davis, M.W., Ailion, M., and Jorgensen, E.M. (2012). Improved Mos1-mediated transgenesis in C. elegans. Nat. Methods 9, 117-118. Abstract Article
Frokjaer-Jensen, C., Davis, M.W., Hopkins, C.E., Newman, B.J., Thummel, J.M., Olesen, S.P., Grunnet, M., and Jorgensen, E.M. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375-1383. Abstract Article
Kelly, W.G., Xu, S., Montgomery, M.K., and Fire, A. (1997). Distinct requirements for somatic and germline expression of a generally expressed Caernorhabditis elegans gene. Genetics 146, 227-238. Abstract
Lo, D.C., McAllister, A.K., and Katz, L.C. (1994). Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron 13, 1263-1268. Abstract Article
Lorence, A., and Verpoorte, R. (2004). Gene transfer and expression in plants. Methods Mol. Biol. 267, 329-350. Abstract
Maduro, M., and Pilgrim, D. (1996). Conservation of function and expression of unc-119 from two Caenorhabditis species despite divergence of non-coding DNA. Gene 183, 77-85. Abstract Article
Merritt, C., Gallo, C.M., Rasoloson, D., and Seydoux, G. Transgenic solutions for the germline (February 8, 2010), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.148.1, http://www.wormbook.org.
Praitis, V. (2006). Creation of transgenic lines using microparticle bombardment methods. Methods Mol. Biol. 351, 93-107. Abstract
Praitis, V., Casey, E., Collar, D., and Austin, J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157, 1217-1226. Abstract
Semple, J.I., Biondini, L., and Lehner, B. (2012). Generating transgenic nematodes by bombardment and antibiotic selection. Nat. Methods 9, 118-119. Abstract Article
Stiernagle, T. Maintenance of C. elegans (February 11, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.101.1, http://www.wormbook.org.
*Edited by Oliver Hobert Last revised November 26, 2012, Published December 30, 2013. This chapter should be cited as: Schweinsberg P.J., Grant B.D. C. elegans gene transformation by microparticle bombardment (December 30, 2013), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.166.1, http://www.wormbook.org.
Copyright: © 2013 Peter J. Schweinsberg and Barth D. Grant. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Email: grant@biology.rutgers.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.