Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
|
‘Nothing in biology makes sense except in the light of evolution’ |
||
| --Theodosius Dobzhansky (The American Biology Teacher, 1973) | ||
Satellite systems are proving to be important players in understanding the differences between ontogenies during evolution. The satellite system P. pacificus, since its discovery nearly a decade ago (Sommer et al., 1996), has provided developmental biologists with new insights into various developmental processes (Eizinger and Sommer, 1997; Jungblut and Sommer, 1998; Sigrist and Sommer, 1999; Zheng et al., 2005). Further understanding of these developmental processes necessitated a genomic framework. Generation of an integrated genetic linkage map and a physical map for mapping of mutants has set a good foundation for such molecular studies (Srinivasan et al., 2003; Srinivasan et al., 2002). With the impending genome sequence being generated by the Washington Genome sequencing center (WUGSC) funded by NHGRI, P. pacificus is bound to cross the ‘next frontier’ in our understanding of evolution of developmental and behavioral processes. This review highlights the current status of P. pacificus genomics.
Pristionchus pacificus is a free-living nematode belonging to the family Diplogastridae. It is 1 mm long and has two sexes viz. self-fertilizing hermaphrodites and cross fertilizing males, making it amenable to genetic analysis. Adult hermaphrodites are larger than males. In P. pacificus, like C. elegans, animals with two X chromosomes develop as hermaphrodites, whereas XO animals develop as males. Males are rare, occurring at a low frequency, but can be induced under low food or stress conditions. The life cycle of P. pacificus at 20°C is 4 days (Sommer et al., 1996). A single self-fertilizing hermaphrodite lays on average 190 eggs over a period of several days. P. pacificus, has the four larval stages seen in many different nematodes including C. elegans. However, we observe a divergence in hatching time with respect to the molt between these species, suggesting that such heterochronic changes play a major role in developmental evolution (Felix et al., 1999). The potential of P. pacificus as a genetic model system has been proven by the generation of a diverse range of mutants affecting several developmental processes such as vulva development, sex determination and gonad development (Pires-daSilva and Sommer, 2004; Rudel et al., 2005; Sommer et al., 1996).
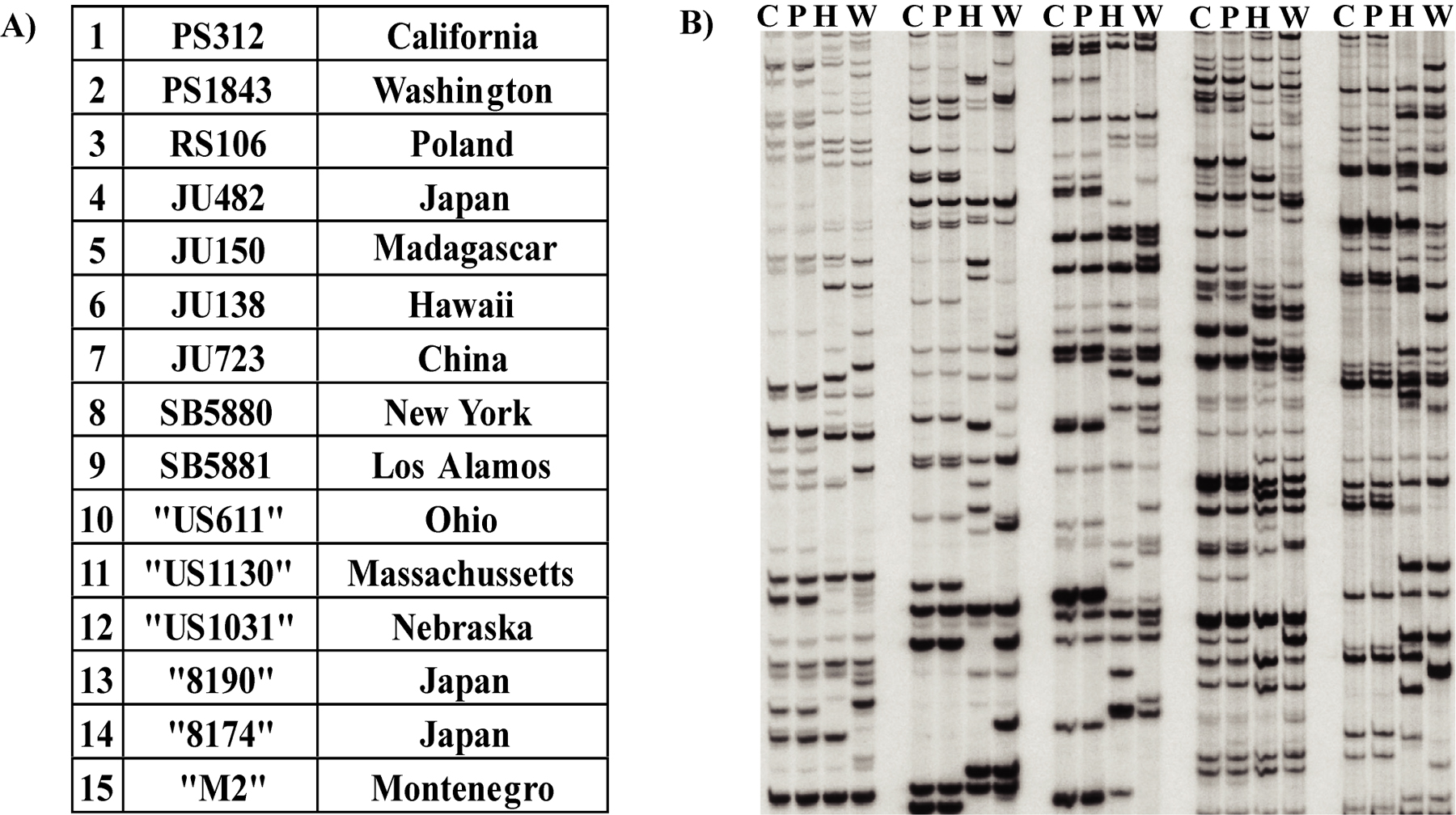
P. pacificus is highly cosmopolitan in distribution and has been isolated from several continents. At present, there are about 15 isolates of P. pacificus (Figure 1). The two commonly used P. pacificus strains PS312 and PS1843 are available at the CGC (http://wormbase.org/db/gene/strain). The other strains can be requested from the Sommer Lab in Germany (http://www.eb.tuebingen.mpg.de/dept4/home.html). Of these, the North American strains, viz. California (PS312), Washington (PS1843), and Hawaii (JU138), and the European strain RS106 from Poland, have identical ITS sequences, representing different isolates from Pristionchus pacificus (Srinivasan et al., 2001; Figure 1). This data was further confirmed by mating experiments between these four species. Males from any of these strains can be mated with hermaphrodites of the other strains (Srinivasan et al., 2001).
Amplified restriction fragment length polymorphism (AFLP) using multiple primer combinations and restriction fragment length polymorphism studies confirmed that the three American strains of P. pacificus differ substantially from one another (Schlak, 1997; Srinivasan et al., 2001). However, the developmentally distinct strain RS106 from Poland exhibited an AFLP pattern that is identical to the one from P. pacificus var. California (PS312) (Srinivasan et al., 2001). Sequence analysis of the polymorphisms indicated that between the polymorphic strains of P. pacificus (PS312 and PS1843), 73.7% were substitutions and 26.3% were insertions/deletions. Of the substitutions, 47.3% were transitions and 52.7% were transversions, a finding that is exactly converse to that of C. elegans (Koch et al., 2000; Srinivasan et al., 2002; Wicks et al., 2001). Data from these polymorphisms provided the necessary prerequisite for further mapping studies to clone genes defined by mutations.
To get a more complete idea of the genome of P. pacificus, a combination of different libraries were generated:
EST library: Initially a mixed-stage library of P. pacificus var. California (PS312) was generated and later on stage-specific libraries were made and sequenced from the oligodT ends (http://www.nematode.net). A total of 14,663 sequences were generated which were then clustered into 2603 clusters. The EST data are accessible at the following links (http://www.ncbi.nlm.nih.gov/dbEST, http://www.nematode.net).
BAC library: Two P. pacificus var. California BAC libraries were generated in collaboration with KeyGene NV Netherlands: one library was constructed by partial digestion of genomic DNA with HindIII and the other was constructed with EcoRI. The average size of the BAC clones from the HindIII library was 126 kb and that from the EcoRI library was 70 kb. Around 700 bp from the 5′ and 3′ ends of each BAC was sequenced (end sequences) at the Genome Centre at the Max Planck Institute for Developmental Biology, Tuebingen. The end sequences were checked for repeats and were blasted against the non-redundant database of NCBI and the C. elegans Wormpep databases. There are currently 31,387 BAC end sequences from both the libraries and they are accessible at NCBI (http://www.ncbi.nlm.nih.gov/entrez).
Fosmid library: A fosmid library of P. pacificus var. California (PS312) was generated containing 50,000 clones in collaboration with California Institute of Technology, Pasadena. From the 50,000 clones, 25,000 clones were end sequenced from both ends at the Genome Sciences Centre, Vancouver, Canada. On average, 500 bp were sequenced from the fosmid ends. 48,568 fosmid end sequences are accessible at NCBI (http://www.ncbi.nlm.nih.gov/entrez).
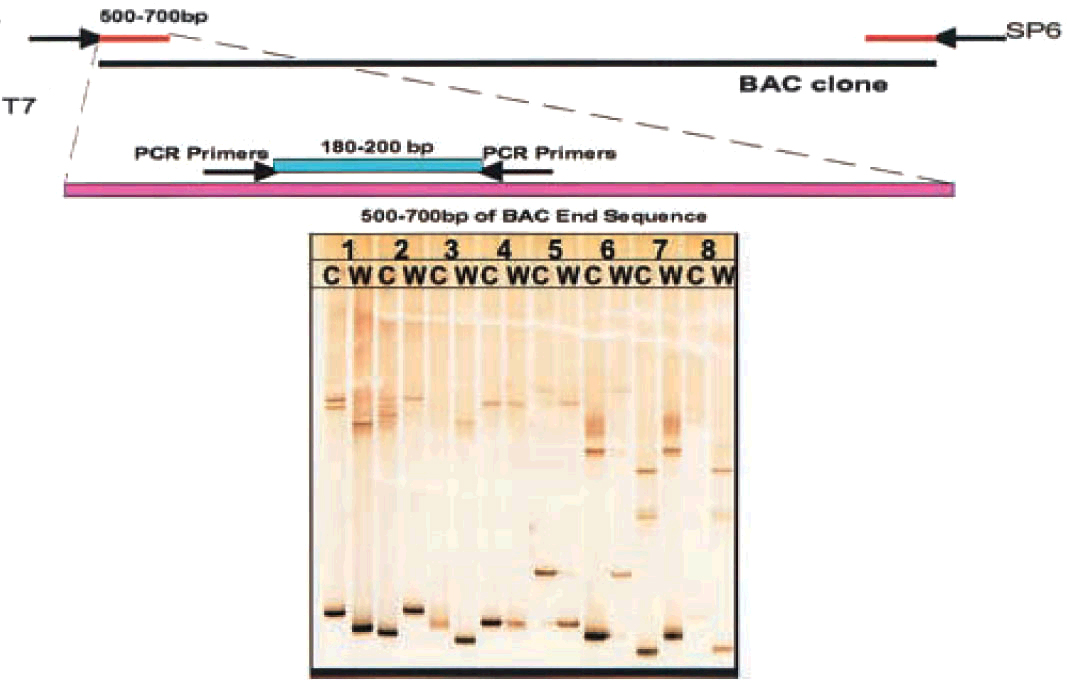
To construct a genetic linkage map, the sequence information from the BAC ends was used to search for polymorphisms between the working ‘wild type’ P. pacificus var. California and the polymorphic Washington strain. We used the “single stranded conformational polymorphism” (SSCP) technique (Orita et al., 1989a), to generate polymorphic markers and map them on the meiotic mapping panel.
The strategy involved designing PCR primers to randomly amplify 180 to 200 bp amplicons in sequenced BAC ends and EST sequences using PrimeArray(c) (Raddatz et al., 2001) and analysing the PCR products for polymorphisms using the SSCP technique (Figure 2; Orita et al., 1989b). As of now we have generated 577 SSCP markers.
|
Figure 2. Strategy for SNP detection: PCR primers were designed using the program PrimeArray to amplify a 180- to 200-bp amplicons (Raddatz et al., 2001). The amplicons were tested for polymorphisms using the SSCP technique. Reprinted with permission from Srinivasan et al. (2002). Copyright © 2002 the Genetics Society of America.
Genomic DNA was extracted from P. pacificus var. California and P. pacificus var. Washington strains using the BioRad genomic DNA extraction kit (BioRad). For a typical PCR assay, 50 ng DNA was used as a template in the following reaction:
| Distilled Water | 6.95 μl |
| Forward Primer (20 μM) | 0.5 μl |
| Reverse Primer (20 μM) | 0.5 μl |
| 10X PCR Buffer | 1.0 μl |
| dNTPs (2 mM) | 1.0 μl |
| Taq Polymerase | 0.05 μl |
Thermocycling was done in a Perkin Elmer Gene Amp9700 PCR machine under standard conditions consisting of an initial denaturation at 94°C for 3 min, followed by 30 cycles of 94°C for 1 min, 50°C for 1 min, and 72°C for 1 min and a final incubation at 72°C for 7 min. The PCR products were checked on a 1% agarose gel for the right size before loading them onto the SSCP gels.
For SSCP detection (Figure 2), the samples were diluted 1:1 in denaturing solution (95% formamide, 0.1% xylene cyanol, 0.1% bromophenol blue, 10mM NaOH), denatured at 95°C for 5 min and loaded onto a GeneGel Excel prepoured 6% acrylamide gel (Pharmacia Biotech). Gels were run for 10 min at 200V, 10 mA, 5W and subsequently 2.45 hours at 375 V, 15mA, 10W. After running, the gels were fixed for 30 min and then stained for 30 min. The gels were washed in water for a minute and then treated with developing solution for 6 min and 30 min in stopping solution to detect the DNA. Mobility differences between the California and Washington PCR products were checked both at the single and double stranded DNA level.
The meiotic mapping panel was prepared from 46 randomly picked F2 animals from a cross between phenotypically marked hermaphrodites of the California strain and Washington strain males. F2 animals were cloned and genomic DNA extracted from them. The polymorphic markers generated were tested on the mapping panel and the markers were assigned linkage groups using the Map Manager Software using the Kosambi mapping function (http://www.mapmanager.org; Manly et al., 2001). Local order was then determined by manually placing the marker at the location that minimised the number of double recombinants. Data analysis ordered 512 loci of the 577 loci into six linkage groups that were supported by LOD scores ≥5.
To assign mutants to chromosomes on the genetic linkage group, two representative markers per linkage group were chosen for analyses. The chosen SSCP markers were from the two opposite ends of each linkage group. Homozygous mutant animals, generated in the California background, were crossed with wild-type Washington strain males and the heterozygous F1 progeny was selfed. DNA from these 21 homozygote animals was isolated and tested for segregation on the representative markers. In order to assign a linkage group, the selected chromosomal markers were tested on these 21 clones and the segregation of the paternal Washington versus the maternal California pattern was tested. The segregation was measured, as a unitless map ratio Washington/California (W/C).
Unlinked markers are expected to be represented equally and henceforth will have a ratio of approximately 1. Linked markers on the other hand, will have a predominant maternal segregation pattern and therefore will yield a value close to 0. To further fine map the mutants, the SSCP markers near to the chromosomal marker showing the least Washingtion/California ratio were tested for segregation with the selfed progeny. The current version of the genetic linkage map can be seen at http://www.pristionchus.org/cgi-bin/cmap.pl.
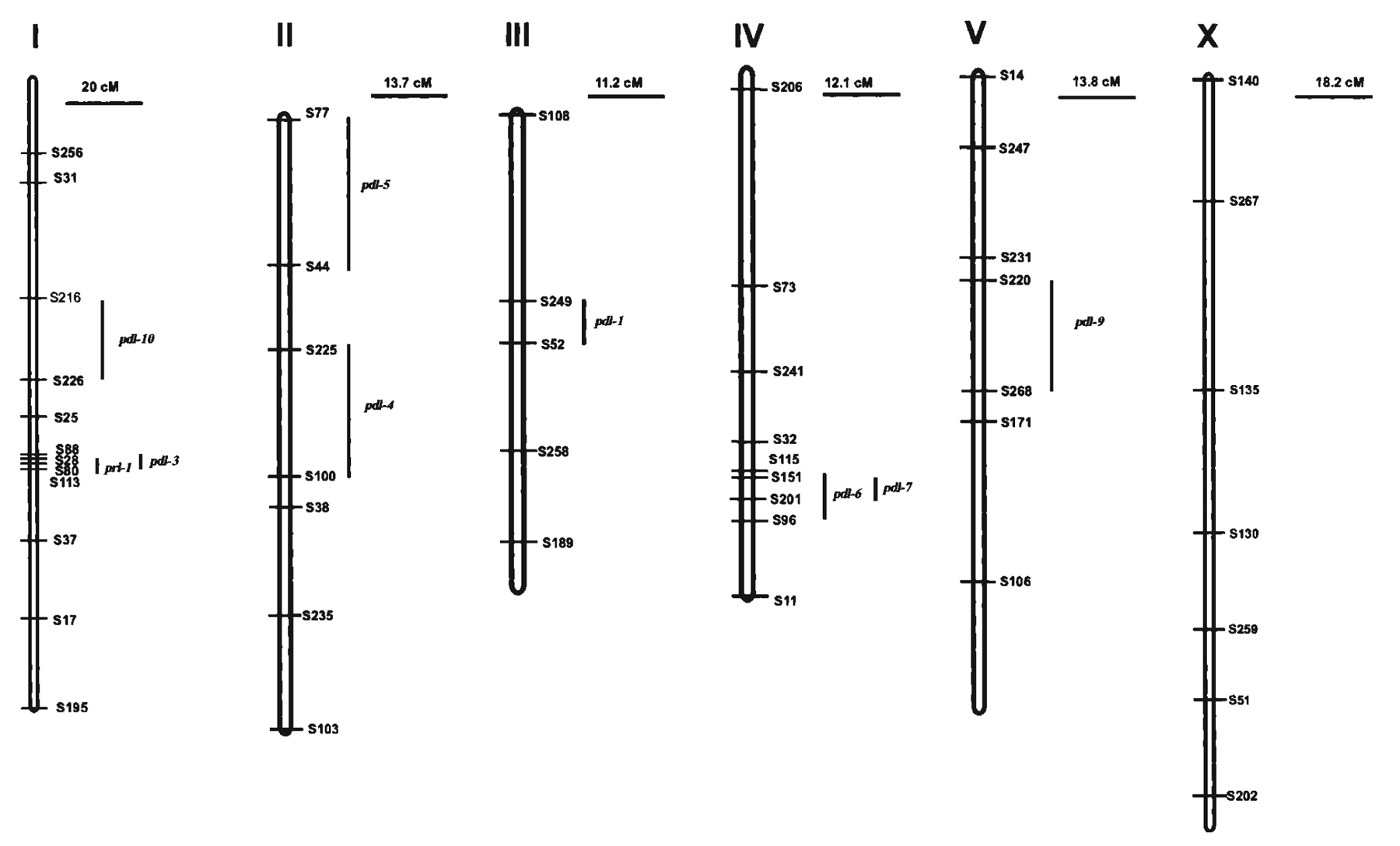
The genetic linkage map of P. pacificus has six chromosomes with five autosomes and one sex chromosome (Figure 3). Sequence similarity comparisons between P. pacificus and C. elegans at a global scale suggest that the six chromosomes can be homologized between the two species (Hong and Sommer, 2006). However, unlike C. elegans, the lengths of the linkage groups exceeds more than 50 cM in length in P. pacificus (Hong and Sommer, 2006). The reason for the increased chromosome length is due to the high incidence of double crossovers per chromosome in P. pacificus compared to one crossover in C. elegans (Hodgkin, 1988; Srinivasan et al., 2002). Molecular studies indicated many intra- but only limited inter-chromosomal rearrangements between the genomes of both species. Additionally, limited microsynteny is seen within a contiguous 126 kb genomic region limiting the likelihood of identifying orthologous genes of P. pacificus and C. elegans based on positional information within the two genomes (Lee et al., 2003; Srinivasan et al., 2002).
|
Figure 3. Genetic linkage map of P. pacificus showing the different linkage groups. The lengths of the linkage groups are not 50cM like C. elegans, due to multiple crossovers.
Mix for 4 Primers (50 μM):
| 1 sample | 26 samples | 28 samples | 55 samples | |
| Distilled Water | 10.2μl | 265.2μl | 285.6μl | 561μl |
| Primer 1 | 0.4μl | 10.4μl | 11.2μl | 22μl |
| Primer 2 | 0.4μl | 10.4μl | 11.2μl | 22μl |
| Primer 3 | 0.4μl | 10.4μl | 11.2μl | 22μl |
| Primer 4 | 0.4μl | 10.4μl | 11.2μl | 22μl |
| 10x PCR Buffer | 2.0μl | 52.0μl | 56.0μl | 110μl |
| dNTPs 2mM | 2.0μl | 52.0μl | 56.0μl | 110μl |
| Taq Polymerase | 0.2 μl | 5.2μl | 5.6μl | 11μl |
Mix for 2 Primers (50 μM):
| 1 sample | 26 samples | 28 samples | 48 samples | |
| Distilled Water | 11μl | 286μl | 308μl | 528μl |
| Primer 1 | 0.4μl | 10.4μl | 11.2μl | 19.2μl |
| Primer 2 | 0.4μl | 10.4μl | 11.2μl | 19.2μl |
| 10x PCR Buffer | 2.0μl | 52.0μl | 56μl | 96.0μl |
| dNTPs 2mM | 2.0μl | 52.0μl | 56μl | 96.0μl |
| Taq Poymerase | 0.2μl | 5.2μl | 5.6μl | 9.6 μl |
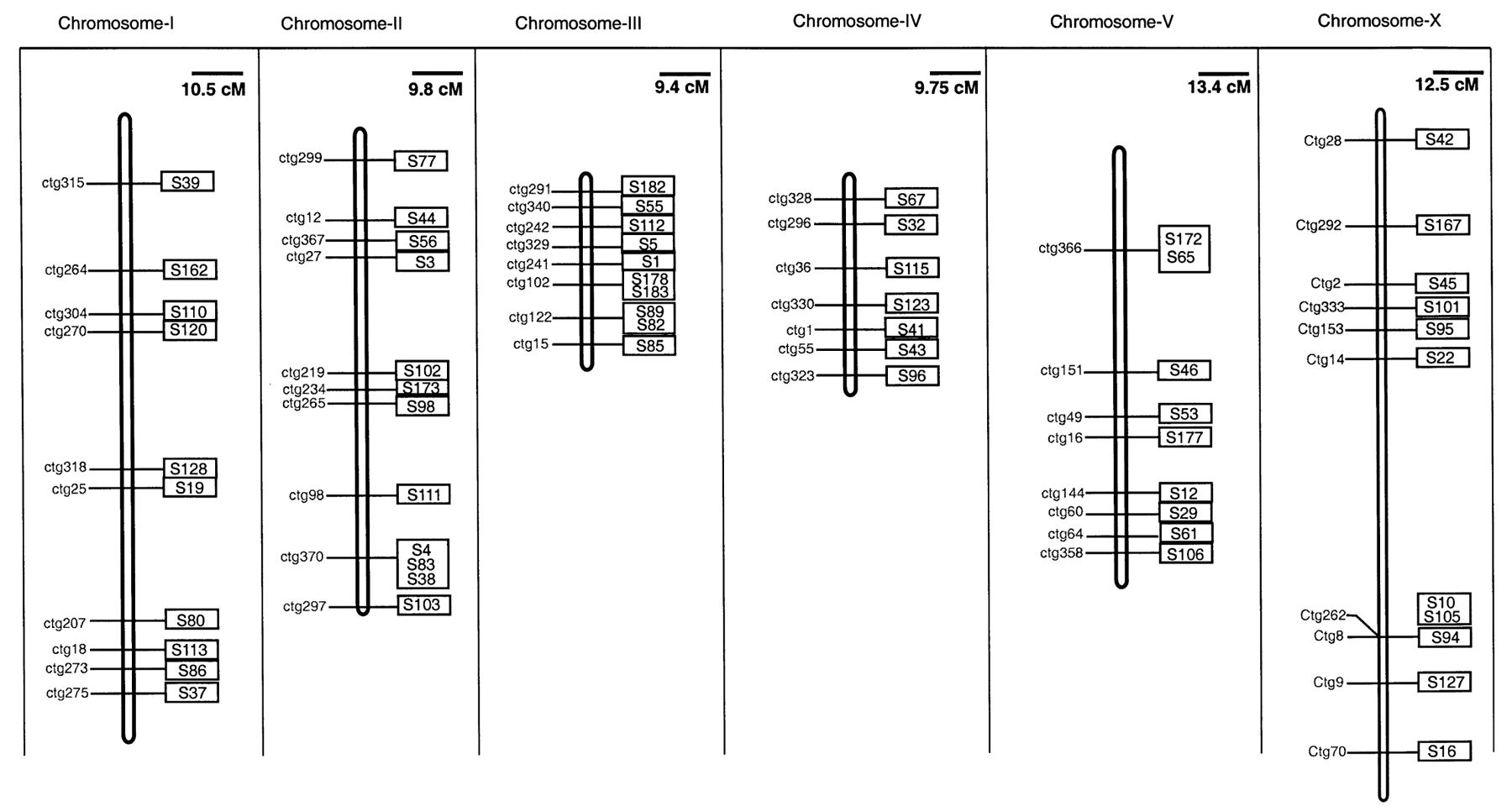
In addition to the molecular SSCP markers, we have isolated 40 morphological markers which can be used to map mutations to the respective chromosomes (Kenning et al., 2004; Figure 4). The morphological markers were isolated in screens for mutations altering body morphology and were classified as ‘Dumpy’, ‘Roller’ and ‘Uncoordinated’ based on their morphological phenotypes. To date, we have isolated 43 Dumpy mutations representing 12 genes and one roller gene. Though the screen is far from being saturated, these genes represent markers for each of the P. pacificus chromosomes. For nomenclature purposes, these dumpy and roller genes are described as Ppa-pdl-n or Ppa-prl-n, where Ppa indicates the species ‘P. pacificus’ and ‘pdl and prl’ stands for Pristionchus dumpy-like and Pristionchus roller-like respectively (Kenning et al., 2004; Sommer et al., 1996).
|
Figure 4. Morphological markers for each linkage group in P. pacificus allowing mapping of mutations in P. pacificus. Reprinted with permission from Kenning et al. (2004). Copyright 2004, John Wiley and Sons.
The physical map of the P. pacificus genome was generated by AFLP fingerprint analysis of BAC clones. Since most of the SSCP markers used to generate the genetic linkage map were derived from BAC ends, the physical genome map and the genetic map could be integrated.
To generate a physical map of the genome of P. pacificus, 9000 BAC clones from the Hind III library were selected, including those BAC clones that had been used to generate the SSCP markers for the genetic linkage map. All the BAC clones were AFLP fingerprinted using the primer combination Hind III/MseI (+0/+0; i.e., no selective bases in the AFLP reaction).
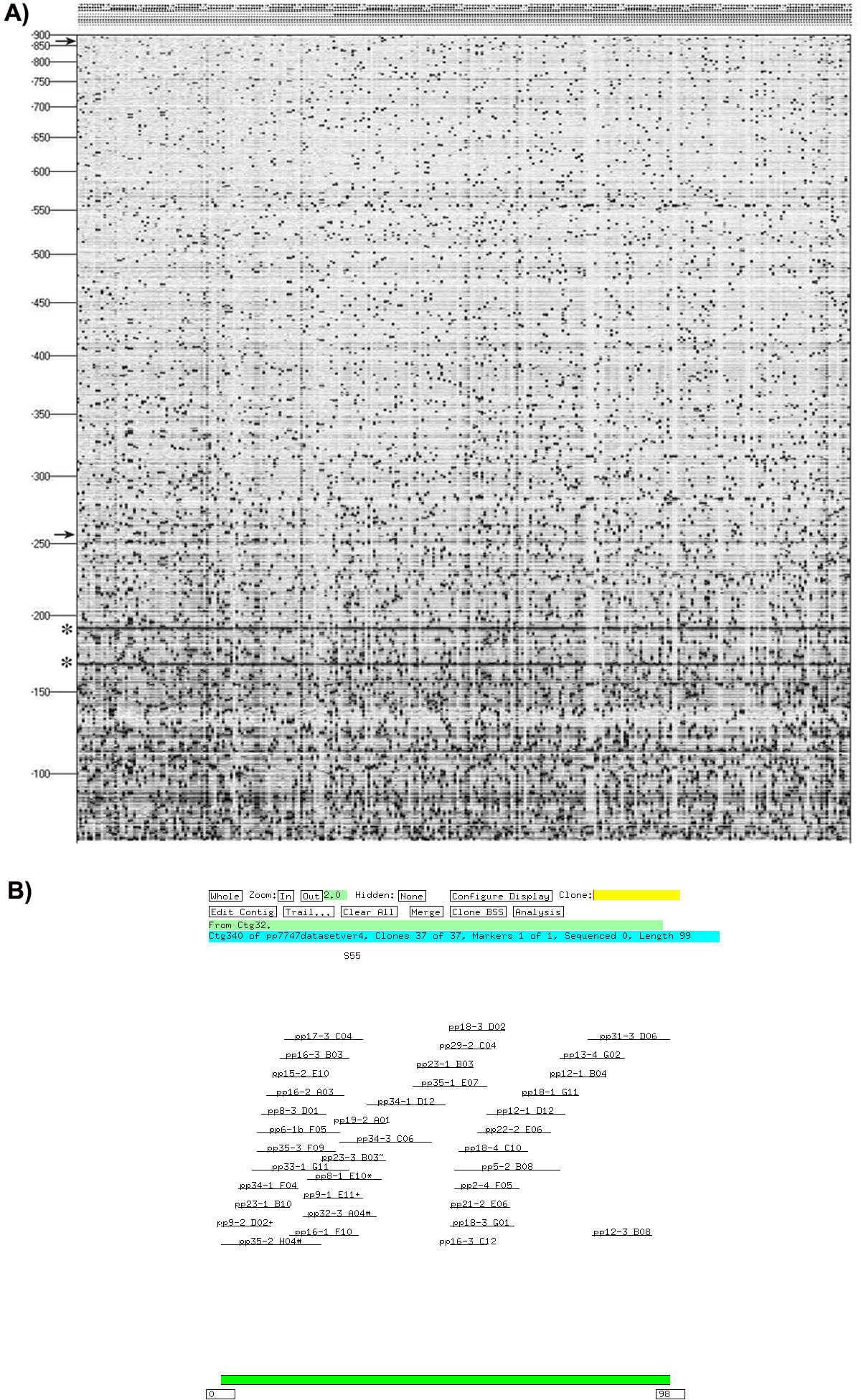
The resulting fingerprints were digitized and scanned for quality control using the program BACxtractor and scanned manually for repetitive elements as these produce excessive numbers of bands (Srinivasan et al., 2003). To optimize contig assembly, a screening window of 260-825 bp was chosen to create the physical map from the AFLP fingerprint data (Figure 5A).
The AFLP based fingerprints for more than 9,000 BAC clones generated were filtered for repeat sequences and bad fingerprinting. On average, 25 AFLP bands per BAC clone were used for fingerprint analysis. Using image analysis, we chose 7747 BAC clones out of the 9,000 clones as suitable for further analysis. These clones were then assembled using FPC v6.2 (Soderlund et al., 1997) to generate the physical map of P. pacificus (Figure 5B).
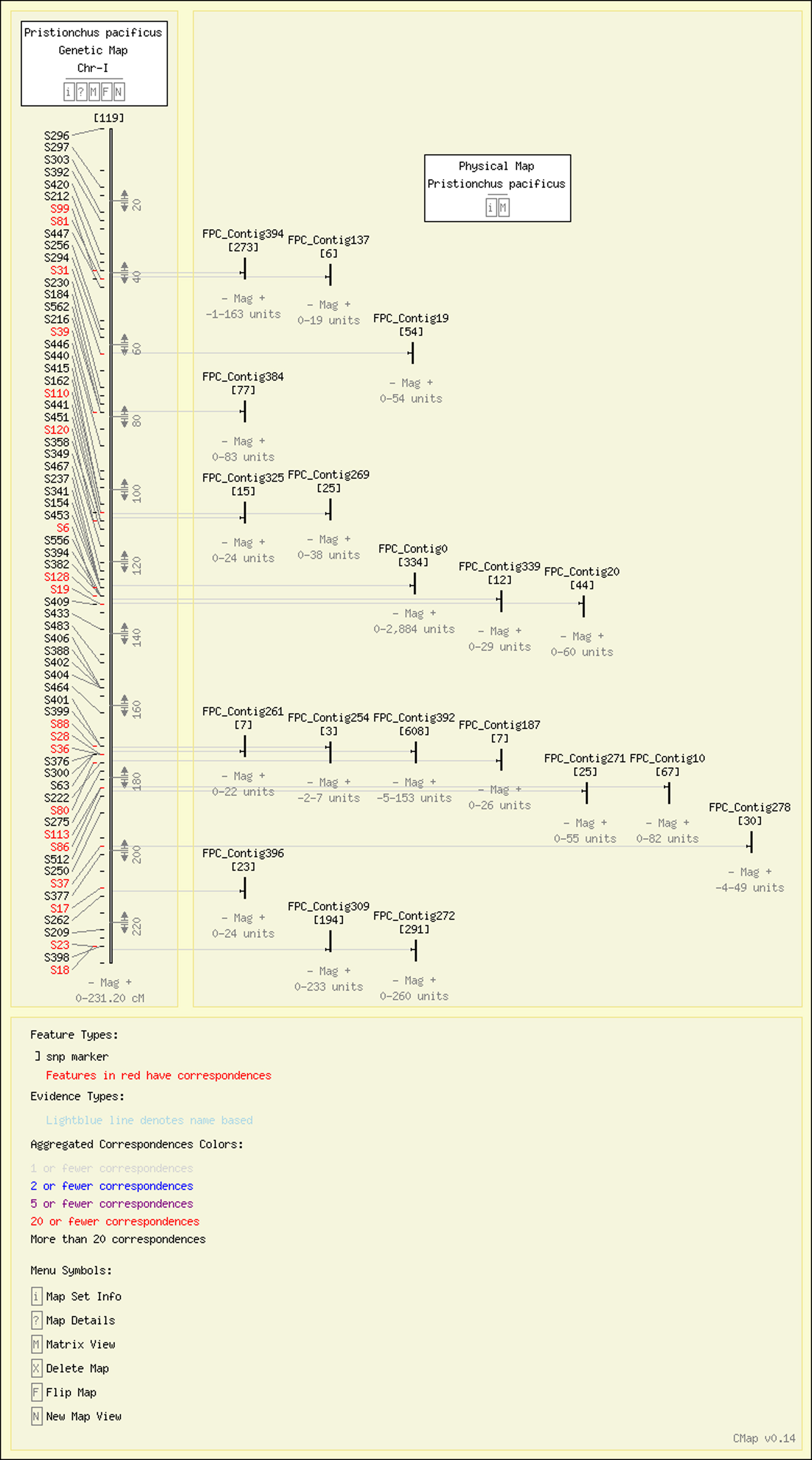
The genetic linkage map of P. pacificus was generated using SSCP markers from BAC-end and EST sequences (Srinivasan et al., 2002) and the physical map of the P. pacificus genome was assembled using AFLP fingerprints of individual BAC clones. This allows many of the SSCP markers on the genetic map can be anchored to the physical map and vice versa. This integration led to 53 contigs containing one or more SSCP markers on the P. pacificus physical map (Figure 6).
|
Figure 5. A) AFLP fingerprint pattern of individual BAC clones using HindIII/Mse-I primer combination. Each lane represents the fingerprint for a single BAC clone. The arrows at the top and bottom of the gel indicates the region of the fingerprint used for analysis. B) FPC v6.2 contig window displaying a horizontal representation of a representative contig (Soderlund et al., 1997). Reprinted from Srinivasan et al. (2003), Copyright 2003, with kind permission of Springer Science and Business Media.
Two large-scale sequencing efforts catapulted P. pacificus into the Genomic era. First, a high-coverage whole genome-sequencing project has been launched and will be completed in late 2006. Second, P. pacificus is part of a large-scale EST sequencing effort at the WUGSC (www.nematode.net; Wylie et al., 2004). These two projects constitute a steady flow of new genomics data. An integrated user-friendly view on these data plus complementary information such as the Genetic Map and Physical Map is provided via our genome portal: www.pristionchus.org.
Our genome portal is the primary access point for all genome-centred data on P. pacificus. The online resource is divided into six sections:
Genome - This section offers a graphical interface to sequence assemblies of the Pristionchus genome and feature annotation thereof. The Generic Genome Browser environment (Stein et al., 2001) is used for this purpose, as it is well known in the nematode community.
BLAST - Similarity searches and single sequence retrieval is easily done on this part of the website.
Genome Maps - Genetic and physical maps are located here. Both are interlinked and will be lined up with the genome assembly once it is available.
Batch Retrieval - Large user-defined data sets are made available via this interface.
WikiOnchus - An online forum for the Pristionchus community.
People - an overview of labs and people working with/on Pristionchus pacificus.
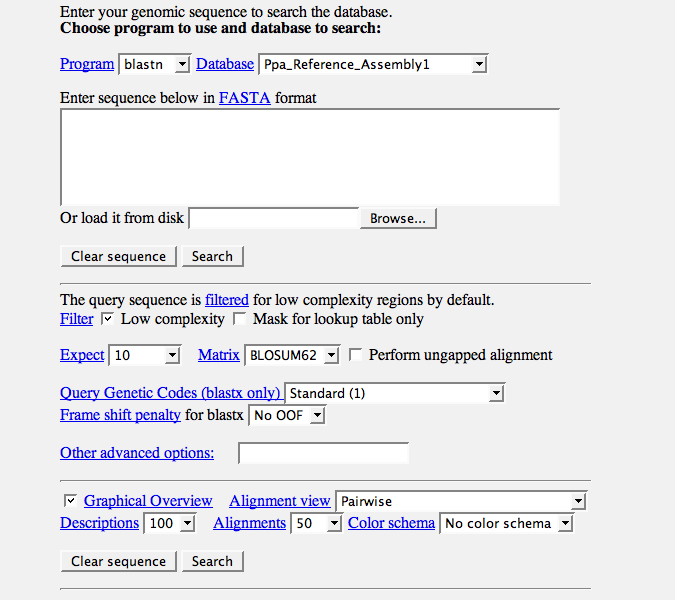
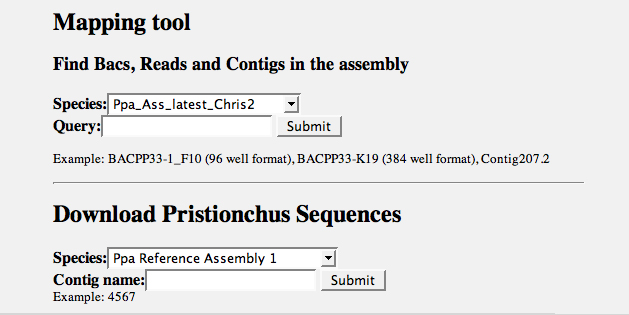
All existing genomic sequence data are deposited in a searchable manner on our website. Genomic sequences can be searched for local similarities with the BLAST form (Figure 7). Search parameters are explained by following the corresponding links. Search parameters can be altered to tailor an individual search approach. Genome assembly constraints are easily checked with the Mapping Tool, which is found below the BLAST form (Figure 7). Here, one obtains information related to genome order, for example like which reads form a Contig and why Contigs are linked into a Supercontig. Single sequence records may be obtained by using the Download tool at the bottom of the page.
We employ the CMap software (Fang et al., 2003) to display our genetic and physical map. The genetic map currently comprises 512 SSCP markers divided up into 6 large linkage groups. The physical map constitutes a second map set encompassing 397 contigs. It was anchored on the genetic map since a subset of genetic markers were derived from BAC ends (Figure 8).
|
Figure 8. Assembly mapping and single sequence download. The Mapping tool delivers information as to the position of a sequence entity on the genome assembly. Individual assembly contigs can be obtained with the Download function. This is particularly useful to retrieve target sequences from BLAST searches.
This section will start to operate with the release of the first whole genome annotation.
WikiOnchus is an online forum on our favourite animal. Everybody is free to view forum contents ranging from methods and protocols to basic introductory texts in a variety of languages. Web users are encouraged to register for active membership to be granted access to edit pages. WikiOnchus is based on the MediaWiki system for which further information are available at http://www.mediawiki.org.
We would like to thank the whole nematode community for their continued interest in a ‘satellite’ model system and Paul W. Sternberg and the anonymous reviewers for critical comments on the manuscript.
Eizinger, A., and Sommer, R.J. (1997). The homeotic gene lin-39 and the evolution of nematode epidermal cell fates. Science 278, 452–455. Abstract Article
Fang, Z., Polacco, M., Chen, S., Schroeder, S., Hancock, D., Sanchez, H., and Coe, E. (2003). cMap: the comparative genetic map viewer. Bioinformatics 19, 416–417. Abstract Article
Felix, M.A., Hill, R.J., Schwarz, H., Sternberg, P.W., Sudhaus, W., and Sommer, R.J. (1999). Pristionchus pacificus, a nematode with only three juvenile stages, displays major heterochronic changes relative to Caenorhabditis elegans. Proc. R. Soc. Lond. B. Biol. Sci. 266, 1617–1621. Abstract Article
Hodgkin, J. (1988). Sexual dimorphism and sex determination. In The nematode Caenorhabditis elegans., W. Wood, ed., Cold Spring Harbor Laboratory Press, pp. 243–279.
Hong, R.L., and Sommer, R.J. (2006) Pristionchus pacificus: a well-rounded nematode. Bioessays 28, 651–659. Abstract Article
Jungblut, B., and Sommer, R.J. (1998). The Pristionchus pacificus mab-5 gene is involved in the regulation of ventral epidermal cell fates. Curr. Biol. 8, 775–778. Abstract Article
Kenning, C., Kipping, I., and Sommer, R.J. (2004). Isolation of mutations with dumpy-like phenotypes and of collagen genes in the nematode Pristionchus pacificus. Genesis 40, 176–183. Abstract Article
Koch, R., van Luenen, H.G., van der Horst, M., Thijssen, K.L., and Plasterk, R.H. (2000). Single nucleotide polymorphisms in wild isolates of Caenorhabditis elegans. Genome Res. 10, 1690–1696. Abstract Article
Lee, K.Z., Eizinger, A., Nandakumar, R., Schuster, S.C., and Sommer, R.J. (2003). Limited microsynteny between the genomes of Pristionchus pacificus and Caenorhabditis elegans. Nucleic Acids Res. 31, 2553–2560. Abstract Article
Manly, K.F., Cudmore, R.H., Jr., and Meer, J.M. (2001). Map Manager QTX, cross-platform software for genetic mapping. Mamm. Genome 12, 930–932. Abstract Article
Orita, M., Iwahana, H., Kanazawa, H., Hayashi, K., and Sekiya, T. (1989a). Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc. Natl. Acad. Sci. U.S.A. 86, 2766–2770. Abstract
Orita, M., Suzuki, Y., Sekiya, T., and Hayashi, K. (1989b). Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5, 874–879. Abstract Article
Pires-daSilva, A., and Sommer, R.J. (2004). Conservation of the global sex determination gene tra-1 in distantly related nematodes. Genes Dev. 18, 1198–1208. Abstract Article
Raddatz, G., Dehio, M., Meyer, T.F., and Dehio, C. (2001). PrimeArray: genome-scale primer design for DNA-microarray construction. Bioinformatics 17, 98–99. Abstract Article
Rudel, D., Riebesell, M., and Sommer, R.J. (2005). Gonadogenesis in Pristionchus pacificus and organ evolution: development, adult morphology and cell-cell interactions in the hermaphrodite gonad. Dev. Biol. 277, 200–221. Abstract Article
Schlak, I., Eizinger,A., and Sommer, R.J. (1997). High rate of restriction fragment length polymorphisms between two populations of the nematode Pristionchus pacificus (Diplogastridae). J. Zool. Syst. Evol. 35, 137–142.
Sigrist, C.B., and Sommer, R.J. (1999). Vulva formation in Pristionchus pacificus relies on continuous gonadal induction. Dev. Genes Evol. 209, 451–459. Abstract Article
Soderlund, C., Longden, I., and Mott, R. (1997). FPC: a system for building contigs from restriction fingerprinted clones. Comput. Appl. Biosci. 13, 523–535. Abstract
Sommer, R.J., Carta, L.K., Kim, S.Y., and Sternberg, P.W. (1996). Morphological, genetic and molecular description of Pristionchus pacificus sp. n. (Nematoda: Neodiplogastridae). Fundam. Appl. Nematol. 19, 511–521.
Srinivasan, J., Pires-daSilva, A., Gutierrez, A., Zheng, M., Jungblut, B., Witte, H., Schlak, I., and Sommer, R.J. (2001). Microevolutionary analysis of the nematode genus Pristionchus suggests a recent evolution of redundant developmental mechanisms during vulva formation. Evol. Dev. 3, 229–240. Abstract Article
Srinivasan, J., Sinz, W., Jesse, T., Wiggers-Perebolte, L., Jansen, K., Buntjer, J., van der Meulen, M., and Sommer, R.J. (2003). An integrated physical and genetic map of the nematode Pristionchus pacificus. Mol. Genet. Genomics 269, 715–722. Abstract Article
Srinivasan, J., Sinz, W., Lanz, C., Brand, A., Nandakumar, R., Raddatz, G., Witte, H., Keller, H., Kipping, I., Pires-DaSilva, A. et al. (2002). A Bacterial Artificial Chromosome-Based Genetic Linkage Map of the Nematode Pristionchus pacificus. Genetics 162, 129–134. Abstract
Stein, L., Sternberg, P., Durbin, R., Thierry-Mieg, J., and Spieth, J. (2001). WormBase: network access to the genome and biology of Caenorhabditis elegans. Nucleic Acids Res. 29, 82–86. Abstract Article
Wicks, S.R., Yeh, R.T., Gish, W.R., Waterston, R.H., and Plasterk, R.H. (2001). Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 28, 160–164. Abstract Article
*Edited by Ralf J. Sommer. WormMethods editor, Victor Ambros. Last revised May 16, 2006. Published August 14, 2006. This chapter should be cited as: Dieterich C., et al. Pristionchus pacificus genomics: from genetics to genome sequence (August 14, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.116.1, http://www.wormbook.org.
Copyright: © 2006 Christoph Dieterich et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: christoph.dieterich@tuebingen.mpg.de or jsrini@caltech.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.