Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
The nematode Caenorhabditis elegans relies on its innate immune defenses to counter infection. In this review, we focus on its response to infection by bacterial and fungal pathogens. We describe the different families of effector proteins that contribute to host defense, as well as the signal transduction pathways that regulate their expression. We discuss what is known of the activation of innate immunity in C. elegans, via pathogen recognition or sensing the damage provoked by infection. Damage causes a stress response; we review the role of stress signaling in host defense to infection. We examine examples of inter-tissue communication in innate immunity and end with a survey of post-transcriptional regulation of innate immune responses.
The maintenance of cellular and organismal homeostasis in the face of changes in the environment is essential for survival. Pathogenic microorganisms represent one type of environmental challenge. Plants and animals have developed immune systems to counter the threat of infection. The innate immune system is an ancient system of host defense against microbial pathogens present across a very broad range of species. Genetic studies of innate immunity in Drosophila and mammals have revealed an evolutionary conservation of signaling mechanisms (Medzhitov and Janeway, 1998), which has been corroborated in part by the study of innate immunity in Caenorhabditis elegans. Because infection can reduce fecundity or cause death, these immune systems are under strong selective pressure from co-evolving microbes. As a consequence, in addition to shared elements of defense signaling pathways, every species also exhibits lineage-specific innovations and losses to their innate immune mechanisms (Ausubel, 2005; Du Pasquier, 2005; Dishaw et al., 2012; Zhang et al., 2015).
This chapter will deal primarily with the signaling pathways that control innate immunity to bacterial and fungal infection in C. elegans and their integration with cellular and organismal physiology. This is an area of study that has expanded enormously since the first edition of this chapter (Signaling in the immune response), and we have been selective in the literature that we cite. The reader is referred elsewhere for various related topics not covered here: innate immunity to other pathogens, including natural viruses (Félix et al., 2011; Ashe et al., 2013; Sarkies et al., 2013) and microsporidia (Troemel et al., 2008; Estes et al., 2011), neurobehavioral responses to microbes (e.g., Pujol et al., 2001; Zhang et al., 2005; Pradel et al., 2007; Schulenburg and Ewbank, 2007; Reddy et al., 2009; Maguire et al., 2011; Zhang and Zhang, 2012) and the study of bacterial virulence mechanisms during infection of C. elegans (Ewbank, 2002; Tan, 2002; Alegado et al., 2003; Sifri et al., 2005; Kurz and Ewbank, 2007; Powell and Ausubel, 2008). In addition, comparative studies in other nematodes (e.g., Abad et al., 2008; Rosso et al., 2013; also see references in Sommer and McGaughran, 2013) will not be covered in this chapter.
In their natural environment, nematodes coexist with a diversity of microbial pathogens, including bacteria, fungi, protozoa, and viruses (e.g., (Drechsler, 1941; Barron, 1977; Ahren et al., 2005; Engelmann and Pujol, 2010; Félix and Braendle, 2010; Félix and Duveau, 2012). Infection can occur via one of several routes in C. elegans: the cuticle and epidermis, the uterus, the rectum, or following colonization of the intestine (Figure 1). As there are no dedicated immune cells in C. elegans, it is these different tissues that are responsible for ensuring host defenses.
|
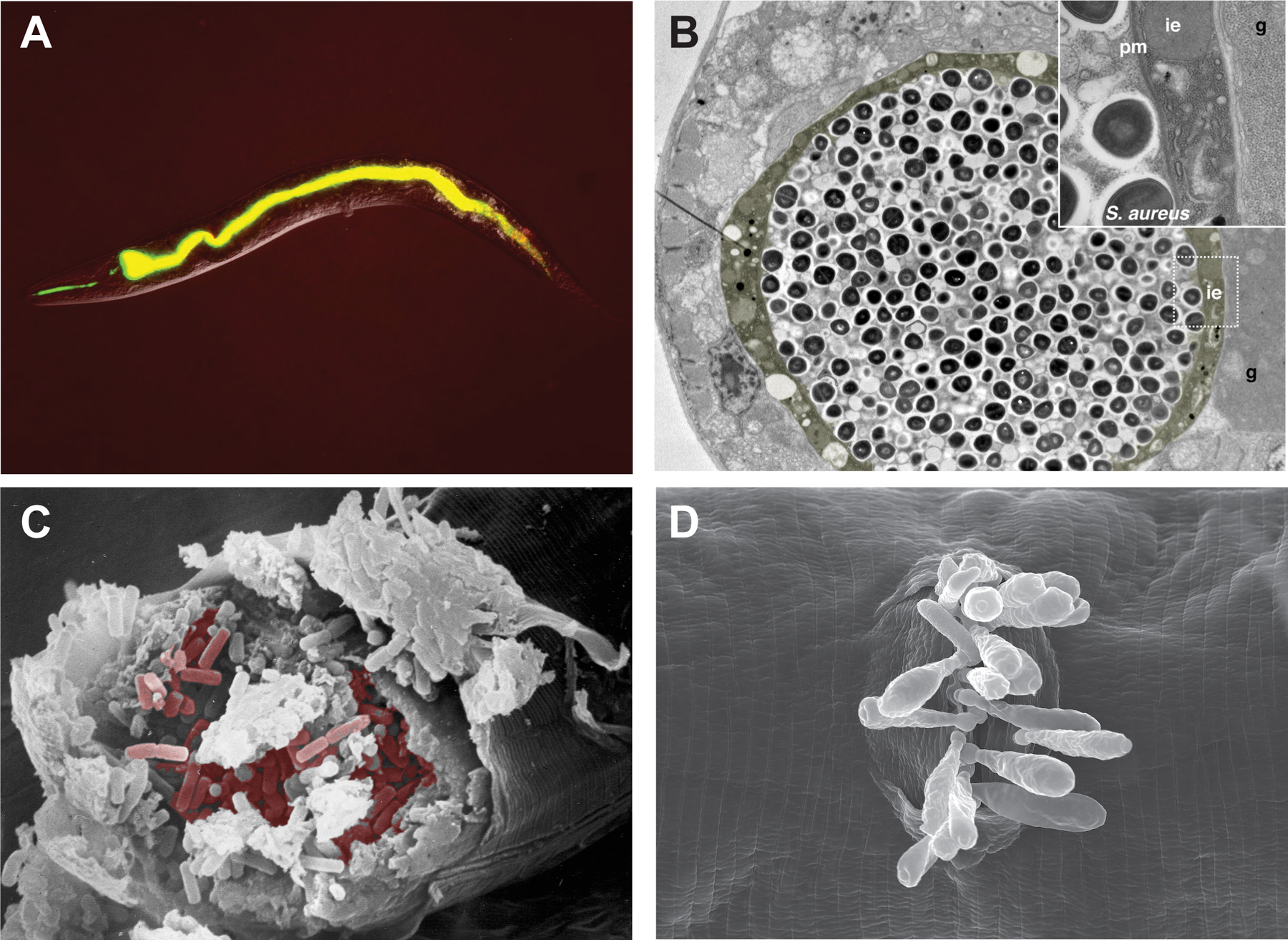
Figure 1. Models of infection in C. elegans. (A) Fluorescence micrograph of an adult hermaphrodite 30 minutes after having been placed on a lawn of Enterococcus faecalis V583::GFP. The intestinal lumen is distended and full of GFP-expressing bacteria. (B) Transmission electron micrograph of an adult hermaphrodite infected by feeding Staphylococcus aureus for 36 h. In this transversal section, the intestinal epithelium (ie, in yellow), and the gonad (g) are highlighted. The inset shows the boxed region at a higher magnification, illustrating the integrity of the plasma membrane (pm) and the dramatic loss of cytoplasmic volume in the intestinal epithelial cell. (C) Scanning electron micrograph of an adult hermaphrodite split open to reveal the large number of Bacillus thuringiensis vegetative cells colonizing the intestine. Some of the B. thuringiensis cells have been false-colored. (D) Scanning electron micrograph of an adult hermaphrodite showing multiple Drechmeria coniospora (strain JUf28) spores attached at the vulva. Original images courtesy of Hakkim Rahamathullah, Fred Ausubel, Javier Irazoqui, Hinrich Schulenburg, and Marie-Anne Félix.
The initial contact between microbe and host may be specific, involving recognition of host surface moieties by the pathogen. Specific mutations in genes encoding surface cuticle proteins or proteins involved in the secretion of the carbohydrate-rich surface coat of the cuticle can reduce this interaction and therefore confer resistance to infection. Examples include mutants of the Bacterially Un-Swollen genes—bus-2, bus-4, bus-12, and bus-17—that limit the attachment of Microbacterium nematophilum to the rectal epithelia (Yook and Hodgkin, 2007; Partridge et al., 2008; Palaima et al., 2010; Gravato-Nobre et al., 2011) and of Yersinia pseudotuberculosis to the head (Cipollo et al., 2004; Hoflich et al., 2004; Drace et al., 2009). In one well-characterized example, loss of function mutants of bus-4, which encodes a glycosyltransferase (Gravato-Nobre et al., 2011), not only have changes in O- and N-glycans, but also marked changes in the expression of the surface mucin genes let-653 and osm-8 (Parsons et al., 2014).
Recent studies using two natural pathogens of C. elegans, both belonging to the bacterial genus Leucobacter (Verde1 and Verde2), have shown that mutations, such as bus-2, bus-4, bus-12, and bus-17, that confer resistance to Verde2 dramatically increase susceptibility to Verde1 (Hodgkin et al., 2013). The fungus Drechmeria coniospora infects these same 4 mutants much more effectively than wild-type worms, because its spores attach more efficiently to the mutants’ cuticle (Rouger et al., 2014). Thus, resistance to some natural pathogens can be associated with increased susceptibility to others. These results reveal the opposing selective forces that must shape the evolution of C. elegans. The varied effects of microbes on host physiology may contribute to the genetic diversity found in nature (Schulenburg and Ewbank, 2004; Barrière and Félix, 2005; Coolon et al., 2009; Andersen et al., 2012; Reynolds and Phillips, 2013).
The cuticle is one of the mechanical barriers that constitute a first line of protection against many potential pathogens. The pharyngeal grinder serves a similar protective role that is illustrated by mutants defective in grinder function. As a consequence of increased ingestion of intact microbes, grinder-defective mutants have enhanced susceptibility to killing by pathogenic bacteria (Labrousse et al., 2000; Kim et al., 2002).
Infection with pathogenic microbes engages the host innate immune system. Innate immunity comprises a number of distinct classes of proteins that mediate the multiple steps involved in a protective response to pathogen infection. First, there are the host receptors that recognize the presence of a pathogen and/or pathogen-induced host damage. Second, signal transduction pathways downstream of the initial recognition of infection require proteins such as kinases and phosphatases, as well as transcriptional regulators that direct changes in gene expression. A phylogenetic comparison of genes encoding components of innate immunity in Drosophila and Anopheles suggested that these core signaling constituents of the innate immune response exhibit the highest degree of structural and functional evolutionary conservation (Christophides et al., 2002), and the lowest level of copy number variation within Drosophila species (Sackton et al., 2007). Third, antimicrobial peptides (AMPs) and proteins as well as secreted signals that coordinate an organismal response to infection (termed cytokines in vertebrates), serve as the effector mechanisms of the innate immune response to control infection. We organize our review by discussing studies of each of these classes of components of C. elegans innate immunity in reverse order, starting with the immune effectors. We go on to discuss how innate immunity is integrated with organismal inter-tissue signaling and stress response pathways in C. elegans.
The secretion of antimicrobial proteins represents an ancient mechanism of innate immunity that is found in both plants and animals. Sequence homology was used to detect the first candidate antimicrobial proteins of C. elegans: the saposin-like amoebapores (a large family of 23 SPP proteins), and AMPs, the defensins, which in C. elegans are called ABF-1 to ABF-6 (AntiBacterial Factor-related). Direct antimicrobial activity has been demonstrated for certain members of both families when produced from recombinant expression systems and tested in vitro (Banyai and Patthy, 1998; Kato et al., 2002; Hoeckendorf et al., 2012) (Table 1). Sequence conservation has also led to the definition of several other families, including lysozymes that may digest peptidoglycans in bacterial cell wall (10 LYS and 6 invertebrate ILYS proteins) (Mallo et al., 2002; Schulenburg and Boehnisch, 2008) and thaumatins, which resemble plant proteins that inhibit fungal growth and sporulation (8 THNs) (Murphy et al., 2003). For many classes of AMPs, function is a consequence of overall structural properties, such as the presence of characteristic amphipathic helices, rather than specific sequence requirements, thus limiting the utility of homology-based approaches for the identification of antimicrobial effectors. Nevertheless, computational approaches based on sequence evolution have been utilized successfully to identify candidate innate immune effectors of C. elegans. For example, on the basis of their predicted structures, the clustering in the genome of the corresponding genes, and the observed concerted sequence evolution, members of the Nematode Specific Peptide group B and C families (encoded by 12 nspb and 20 nspc genes) have been proposed to be AMPs (Thomas, 2006).
Table 1. A selection of C. elegans immune effectors.
| Protein family | Evidencea | Controlled byb | Tissue(s) | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| iv | Tr | Hom | GoF | LoF | p38 | ins | TGF | |||
| ABF | X | X | X | X | X | pharyngeal neurons, marginal and excretory cells | (Kato et al., 2002; McElwee et al., 2004; Ren et al., 2009; Pukkila-Worley et al., 2011) | |||
| CNC | X | X | X | X | epidermis | (Couillault et al., 2004; Pujol et al., 2008b; Zugasti and Ewbank, 2009) | ||||
| CLEC & LEC | X | X | X | X | X | X | X | (Mallo et al., 2002; Murphy et al., 2003; O'Rourke et al., 2006; Alper et al., 2007; Irazoqui et al., 2010) | ||
| ILYS & LYS | X | X | X | X | X | X | X | Intestine, head and tail neurons, rectal gland cells | (Mallo et al., 2002; Alper et al., 2007; Schulenburg and Boehnisch, 2008; Irazoqui et al., 2010; Boehnisch et al., 2011; Marsh et al., 2011) | |
| NLP | X | X | X | X | Epidermis, vulva | (Nathoo et al., 2001; Couillault et al., 2004; Wong et al., 2007; Pujol et al., 2008b; Dierking et al., 2011) | ||||
| SPP | X | X | X | X | X | X | X | pharyngeal muscles and neurons, intestine | (Banyai and Patthy, 1998; Murphy et al., 2003; Alper et al., 2007; Alegado and Tan, 2008; Evans et al., 2008; Roeder et al., 2010; Hoeckendorf et al., 2012) | |
|
a iv.: in vitro antimicrobial activity; Tr: transcriptional change upon infection; Hom: homolog(s) in other species with demonstrated immune effector function; Gof: genetic gain-of-function (e.g., over-expression leads to protection); Lof: genetic loss-of-function (e.g., RNAi leads to susceptibility). b At least one member shown to be controlled by one of the 3 pathways, p38: PMK-1 p38 MAPK pathway; ins: DAF-2/DAF-16 insulin signaling pathway; TGF: DBL-1 TGF-ß pathway. |
||||||||||
Expression profiling methods have also been used to identify candidate immune effector genes. An initial study of C. elegans genes expressed during infection by the bacterial pathogen Serratia marcescens revealed the induction of several lysozyme genes, including lys-1. By inference, many of the other up-regulated genes of unknown function, such as F55G11.4, were assumed to be innate immune effectors (Mallo et al., 2002). This gene encodes a CUB-like (for complement C1r/C1s, Uegf, Bmp1-like; previously called DUF141) domain protein. While the Caenorhabditis-specific CUB-like domain and the structurally-related and phylogenetically widespread CUB domain are present in a wide range of proteins of diverse function (http://ipfam.org/family/PF00431 and http://pfam.xfam.org/family/PF02408), including 50 in C. elegans, its role in nematode defense has yet to be elucidated. The study by Mallo et al. and subsequent genome-wide expression profiling analyses (e.g., O'Rourke et al., 2006; Shapira et al., 2006; Troemel et al., 2006; Wong et al., 2007; Bolz et al., 2010; Engelmann et al., 2011; Pukkila-Worley et al., 2011; Sahu et al., 2012; Sinha et al., 2012) have identified numerous other genes that are induced during infection. Depending on the particular pathogen, these can include some of the 265 C-type lectin (clec) genes, certain galectin genes (from lec-1 to lec-12) (Nicholas and Hodgkin, 2004; Schulenburg et al., 2008), and genes encoding other proteins with CUB or CUB-like domains, as well as members of the Fungus-Induced Protein and FIP-Related gene families (7 fip and 29 fipr genes) that are presumed to encode AMPs (Pujol et al., 2008b; Pujol et al., 2012), and AMPs of the Neuropeptide-like protein (nlp) and Caenacin (cnc) families (Couillault et al., 2004).
These transcriptomic studies have been complemented by a number of proteomic analyses (e.g., Simonsen et al., 2011; Couillault et al., 2012). While earlier studies were not quantitative on the proteome-wide scale, more recently, quantitative analyses of the host response to pathogenic Bacillus thuringiensis and to the Pseudomonas aeruginosa strain PA14 have been published (Treitz et al., 2015; Witting et al., 2015; Yang et al., 2015). A number of classes of proteins have been identified by both methods, most prominently lectins, including LEC-6 (reviewed in Simonsen et al., 2012). Lectins in general, and C-type lectins in particular, have been implicated in innate immunity of diverse species (van den Berg et al., 2012). Their precise role in C. elegans defense has been the subject of speculation for more than a decade (Mallo et al., 2002). In a recent study (Miltsch et al., 2014), it was shown that recombinant CLEC-39 and CLEC-49 can directly bind to S. marcescens. Interestingly, while clec-49 (but not clec-39) has been reported to be induced by S. marcescens infection (Engelmann et al., 2011), both clec-39 and clec-49 mutants have a heightened susceptibility to S. marcescens infection. Unlike certain C-type lectins with direct antimicrobial activity (Vaishnava et al., 2011), however, neither CLEC-39 nor CLEC-49 appear to be bactericidal (Miltsch et al., 2014). Adding to the enigma, in common with many C. elegans C-type lectins (Drickamer and Dodd, 1999; Schulenburg et al., 2008), CLEC-39 is not predicted to be a cell-surface or secreted protein, unlike CLEC-49 that has a clear secretion signal peptide. Two models can be proposed to explain the function of CLEC-49: either it could act during pathogen-recognition and be involved in triggering defense gene expression, or it could act synergistically with bactericidal proteins, such as lysozymes or amoebapores, sensitizing bacteria to their killing action. Examining bacterial loads and defense gene induction in the clec-49 mutant should help distinguish between the two.
Transcriptomic and proteomic analyses thus provide candidate immune effectors. As in the case of the clec genes just described, functional corroboration of a role for candidate genes in innate immunity has been attempted primarily through studies of gene inactivation or over-expression of single or multiple candidate genes being associated with the expected change in resistance. With regard to C. elegans lysozymes, enhanced resistance against S. marcescens was observed for animals over-expressing lys-1 (Mallo et al., 2002), enhanced resistance to Staphylococcus aureus accompanied overexpression of lys-4 and lys-5 together (Irazoqui et al., 2010), and increased resistance to B. thuringiensis was observed upon overexpression of lys-5 and lys-7 (Boehnisch et al., 2011). Conversely, loss of lys-7 activity in mutants or by RNAi increased susceptibility to M. nematophilum infection (O'Rourke et al., 2006). Additional putative antimicrobial proteins and peptides have also been shown to promote resistance to pathogens, including clec-70 and clec-71 (to S. aureus) (Irazoqui et al., 2010), spp-12 (to B. thuringiensis) (Hoeckendorf et al., 2012), and the nlp-29 and cnc-2 clusters of AMP genes (to the fungus D. coniospora) (Pujol et al., 2008b; Zugasti and Ewbank, 2009).
At the same time, genetic redundancy among multiple co-regulated immune effector genes has undoubtedly confounded efforts to validate putative immune effectors identified from gene expression studies. Troemel et al. (2006) chose 38 genes that are induced by intestinal infection with P. aeruginosa PA14 and regulated by the PMK-1 pathway (see Section 3.1) and knocked down their expression by RNAi. Bearing in mind the usual caveats for RNAi, including inefficiency and off-target effects (Thakur et al., 2014), in no case did they see an effect on pathogen susceptibility (Troemel et al., 2006). In another example, having identified 197 and 35 genes that were respectively induced and repressed by PA14 infection, Shapira et al. (2006) assayed the effect on survival of knocking most of them down individually. They found that RNAi against 17 of the induced genes (9%) specifically reduced survival upon infection by pathogenic bacteria. Curiously, a similar proportion of the transcriptionally repressed genes (11%) also compromised pathogen resistance when inactivated by RNAi (Shapira et al., 2006). Such results highlight the challenge of assigning a specific role at the organismal level for any gene in innate immunity purely on the basis of its expression, a subject that is discussed further in Section 5.3.
The possible role of cytokine-like molecules in coordinating C. elegans immunity has been little explored. Although endocrine signaling might be anticipated to have a more critical role in organizing an immune response involving dedicated immune cells, as observed in vertebrates, one could also envision a role for secreted signals in C. elegans coordinating organismal physiology in response to infection. In C. elegans, insulin and TGFβ pathways are best understood in terms of their roles in the neuroendocrine regulation of host defenses in a cell non-autonomous fashion (Zugasti and Ewbank, 2009; Kawli et al., 2010). This is discussed further in Section 6, Inter-tissue signaling in innate immunity. Further secreted signals, such as ascarosides (von Reuss et al., 2012), might mediate inter-animal signaling. Ascarosides, including dauer pheromone, and other uncharacterized small molecules, influence multiple aspects of nematode development, behavior, and physiology, as well as nematode interactions with microbes (Kaplan et al., 2009; Yamada et al., 2010; Artyukhin et al., 2013; Ludewig et al., 2013; Ludewig and Schroeder, 2013; Olofsson, 2014).
In contrast to the functional redundancy evident in the study of immune effectors, the core signal transduction pathways would be expected to be highly amenable to genetic dissection. Indeed, the application of molecular genetic methods to the identification of genes required for survival during infection has allowed the identification of conserved immune signaling pathways. In the three well-characterized systems of infection of C. elegans described below evolutionarily conserved mitogen-activated protein kinase (MAPK) pathways have been found to have a central role in resistance to microbial pathogens (Figure 2). MAPK pathways play conserved signaling roles during development and for the transduction of environmental stimuli to generate cellular responses in evolutionarily diverse organisms. The ancestral Hog1p MAPK pathway in yeast is involved in the response to osmotic stress (Posas and Saito, 1997). In mammals, the p38 and JNK stress-activated MAPKs are pivotal regulators of both innate defenses and responses to abiotic stress (Huang et al., 2009; Keshet and Seger, 2010). Genetic analysis of C. elegans has established ancient, evolutionarily conserved roles for MAPK signaling in innate immunity.
|
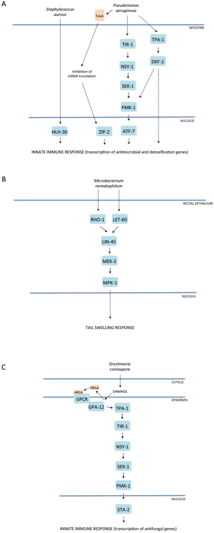
Figure 2. Mitogen-activated protein kinase (MAPK) pathways in C. elegans innate immunity. Evolutionarily conserved MAPK signaling pathways regulate innate immune responses to microbial pathogen infection in C. elegans. Three simplified examples of well-characterized signaling cascades that act in (A) the intestine, (B) rectal epithelium, and (C) epidermis. Alternative representations for A and C can be found at http://www.wikipathways.org/index.php/Pathway:WP2923 and http://www.wikipathways.org/index.php/Pathway:WP2233, respectively.
A forward genetic screen for C. elegans mutants with enhanced susceptibility to killing by pathogenic P. aeruginosa led to the identification of a conserved PMK-1 p38 MAPK pathway. Mutations in genes encoding the upstream activating kinases, nsy-1 and sek-1, as well as in the p38 MAPK gene pmk-1, all conferred enhanced susceptibility to killing by P. aeruginosa, while not affecting survival when worms were propagated on the standard laboratory food source Escherichia coli OP50, which is relatively non-pathogenic. The pivotal role of p38 MAPK signaling in innate immunity in mammals, and specifically downstream of the MAPKKK ASK1, ortholog of NSY-1, immediately suggested that this pathway regulates innate immunity in C. elegans (Kim et al., 2002; Kim et al., 2004). Subsequent studies have implicated this pathway in host resistance to a broad range of bacterial and fungal pathogens, not only in the intestine (e.g., Aballay et al., 2003; Sifri et al., 2003; Bolz et al., 2010; Pukkila-Worley et al., 2011; Muhammed et al., 2012), but also in the epidermis (see Section 3.2).
Whereas genetic studies have established that PMK-1 is required for resistance to killing by bacterial pathogens, the direct detection of an increase in phosphorylated PMK-1 upon exposure to pathogenic bacteria, has not been demonstrated. Irrespective of this, a substantial overlap has been found between genes regulated by the PMK-1 pathway and those induced upon infection by P. aeruginosa (Troemel et al., 2006). The results from this study also suggest an important contribution of the PMK-1 pathway in the regulation of both the basal expression of innate immune effectors and their inducible expression.
A Toll-Interleukin-1 Receptor (TIR) domain adaptor protein TIR-1, which is orthologous to mammalian SARM, functions to activate the PMK-1 pathway in C. elegans innate immunity. SARM is a distantly related member of the family of TIR domain adaptor proteins. The other four mammalian members of this family of signaling proteins, MyD88, TRIF, TIRAP, and TRAM, function to bridge Toll-like Receptors (TLR) to downstream signaling components in innate immunity. In contrast to the TLR-dependent activation of these four adaptor proteins, what activates SARM remains unclear. Indeed, studies of SARM have produced conflicting data regarding its role in innate immunity. Overexpression of SARM in cell culture led to the suggestion that SARM functions as a negative regulator, inhibiting both TRIF- and MyD88-mediated AP-1 activation, and possibly also directly inhibiting MAPK phosphorylation (Peng et al., 2010). More recently, however, it was shown that SARM is required in murine macrophages for the production of the cytokine CCL5. While SARM is not necessary for TLR-induced activation of MAPKs or of transcription factors implicated in CCL5 induction, it is critical for the recruitment of transcription factors and of RNA polymerase II to the Ccl5 promoter (Gurtler et al., 2014). Of note, SARM has also been demonstrated to have a conserved role in promoting neuronal degeneration (Osterloh et al., 2012). In C. elegans, TIR-1 is the only TIR-domain adaptor protein (Pujol et al., 2001), and TIR-1 is required for PMK-1 activation and host resistance to killing by several pathogens (Couillault et al., 2004; Liberati et al., 2004). The mechanisms by which TIR-1 is activated in innate immunity are unclear. Moreover, the single TLR homolog in C. elegans TOL-1 appears not to have a role in intestinal or epidermal innate immunity, nor in PMK-1 activation (Couillault et al., 2004; Irazoqui et al., 2010; Irazoqui et al., 2010).
Diacylglcerol (DAG) levels play an important role in intestinal defenses. Both the protein kinase D DKF-2, and the upstream protein kinase C (PKC) TPA-1, are activated by DAG. DKF-2 contributes to PMK-1 activation, possibly by directly phosphorylating TIR-1. Overexpression of DKF-2 increases PMK-1 phosphorylation and resistance to P. aeruginosa, while dkf-2 mutants exhibit a relatively modest reduction in pathogen resistance (Ren et al., 2009), suggestive of redundancy upstream of TIR-1. It is interesting to note that TIR-1 also has a role in determining an asymmetric neuronal cell fate decision and serotonin biosynthesis in the ADF neuron, but in the nervous system TIR-1 functions downstream of the calcium-calmodulin-dependent kinase UNC-43 (Chuang and Bargmann, 2005; Xie et al., 2013), which appears not to be directly involved in the regulation of innate immunity (Kim et al., 2002; Pujol et al., 2008a). This difference may reflect the fact that the isoforms of TIR-1 appear to have distinct roles in defense or development (Chuang and Bargmann, 2005; Pujol et al., 2008a).
While genetic analyses in mammals have hitherto been limited because of the essential requirement for p38 MAPK in development, biochemical and cell-based approaches have implicated a diverse array of substrates for p38 MAPK in mammalian systems. These include a large number of transcription factors for which direct phosphorylation and activation have been described, such as ATF1, ATF2, CHOP, c/EBPs, CREB, Elk1, MEF2A/C, SAP1A, NFAT, p53, SP1, and STAT1. Although some of these direct p38 MAPK targets are absent from C. elegans, notably NFAT (Sullivan et al., 2009), an ubiquitous regulator of cell differentiation and activation in vertebrates, there are 5 members of the conserved CREB/ATF transcription factor family in worms. Among them, ATF-7 has been shown to function downstream of the PMK-1 pathway in the innate immune response against bacterial pathogens in the C. elegans intestine. The characterization of gain-of-function and loss-of-function alleles of atf-7 has led to a model for PMK-1-ATF-7 function in which ATF-7 serves as a transcriptional repressor that is converted to an activator upon phosphorylation by PMK-1 (Shivers et al., 2010). Such a mechanism parallels that underlying the activation of the yeast Hog1p-Sko1p pathway, underscoring the evolutionarily ancient molecular origins of host defense mechanisms.
Following the attachment of their non-motile spores (conidia) to the nematode cuticle, certain pathogenic fungi, such as D. coniospora, cause invasive infection through the direct extension of hyphae into the epidermis (Dijksterhuis et al., 1990). Infection of C. elegans with D. coniospora has been shown to induce the transcription of many genes encoding potential AMPs, including all six genes of the nlp-29 locus (Pujol et al., 2008b; Engelmann et al., 2011). The use of an nlp-29p::GFP reporter construct has facilitated the genetic identification of genes that regulate the epidermal response to fungal infection and established a framework for innate immune signaling in the epidermis. Interestingly, the same TIR-1-NSY-1-SEK-1-PMK-1 signaling pathway that promotes pathogen resistance to bacteria in the intestine functions in the epidermis to regulate the induction of antifungal peptide gene expression by Drechmeria (Pujol et al., 2008b), as well as upon wounding (Pujol et al., 2008a). Since the model of infection by D. coniospora gives insight into the question of whether and how worms respond to a specific pathogen, rather than to the pathology associated with infection, below we go into detail about the role of the different characterized host signaling components; an overview is provided at http://www.wikipathways.org/index.php/Pathway:WP2233.
In the epidermis, the PKC TPA-1 acts upstream of the TIR-1-NSY-1-SEK-1-PMK-1 pathway and downstream of the Gα and Gβ protein genes gpa-12 and rack-1. The pathway is induced by both fungal infection and physical injury (Ziegler et al., 2009). The death-associated protein kinase DAPK-1 functions as a negative regulator of the TIR-1-NSY-1-SEK-1-PMK-1 pathway in the epidermis, and loss of this negative regulator provokes an inappropriate cellular wound healing response and high constitutive expression of AMP genes (Tong et al., 2009).
The downstream target(s) of PMK-1 signaling in the epidermal response to fungal infection remains unclear. STA-2, a protein with distant homology to STAT-like transcription factors that have pivotal roles in mammalian immune responses, has been shown to be required for both PMK-1-dependent nlp gene expression and the PMK-1-independent induction of a second group of AMP genes, the caenacins (cnc-1 to cnc-11), many of which are also induced by D. coniospora infection (Engelmann and Pujol, 2010). Biochemical assays in a heterologous system indicated that STA-2 is a potential substrate of activated PMK-1, suggesting that STA-2 could be a direct downstream target of the PMK-1 pathway in vivo (Dierking et al., 2011).
SNF-12, a member of the SLC6 family of membrane-bound transporters with unknown substrate specificity, was shown to physically interact with STA-2. Epistasis experiments placed snf-12 and sta-2 downstream of sek-1. In contrast to sek-1 mutants, however, in both snf-12 and sta-2 mutants phosphorylation of PMK-1 was normal, suggesting that SNF-12 and STA-2 act either downstream or in parallel to PMK-1 to regulate AMP expression (Dierking et al., 2011).
A number of additional genes, mainly isolated through genetic screens, have been shown to affect the expression of the nlp-29p::GFP reporter gene in response to infection with Drechmeria. The atypical PKC, PKC-3, which lacks a DAG-binding domain present in archetypal PKCs, is presumed to act upstream of PMK-1. pkc-3 acts non-redundantly with tpa-1, upstream of or in parallel to gpa-12, to control nlp-29 after both infection and injury. Loss of two of the six C. elegans phospholipase C genes, plc-3 or egl-8, that catalyze the production of DAG from phosphatidylinositol biphosphate (PIP2) essentially abolishes the induction of nlp-29p::GFP expression upon fungal infection. Although the response to injury is largely independent of egl-8, RNAi against plc-3 results in a marked decrease in the wound-induced upregulation of reporter gene expression (Ziegler et al., 2009). A single allele of the Tribbles-like gene nipi-3 was isolated in a genetic screen for mutants blocking nlp-29p::GFP expression upon fungal infection. As the nipi-3(fr4) mutant does not block the induction of the reporter gene upon injury, it was proposed to be part of an infection-specific branch of the signaling cascade (Pujol et al., 2008a). More recently, epistasis experiments using a constitutively active form of GPA-12 expressed specifically in the adult epidermis, which provokes a marked increase in the expression of several nlp and cnc genes, have placed nipi-3 downstream of, or in parallel to, gpa-12 (Labed et al., 2012), revealing a more complex regulatory mechanism than was originally envisioned. The BiP/GRP78 homolog hsp-3 has also been shown to be required, via an unknown mechanism, for nlp-29p::GFP expression specifically upon fungal infection (Couillault et al., 2012). These results lend support to the notion of two separable inputs, one from D. coniospora infection and one from physical injury converging upstream of TPA-1, and provide further evidence for an important role for DAG-mediated signaling in epidermal defenses, as in the intestine.
While NIPI-3 appears to be required for both epidermal antifungal immunity and intestinal resistance to bacterial infection (D. Kim and N. Pujol, unpublished data), the protein kinase D DKF-2 is not required for the induction of nlp-29p::GFP expression following D. coniospora infection (Ziegler et al., 2009), even though animals overexpressing a dkf-2p::DKF-2-GFP transgene exhibit a marked increase in the expression of multiple nlp and cnc AMP genes (Ren et al., 2009). Similarly, both snf-12 and sta-2 appear to be dispensable for induction of a number of intestinal defense genes (Dierking et al., 2011), reflecting specialization of innate immune signaling pathways in different tissues.
Observation of hermaphrodite animals with altered tail morphology led to the identification of the Gram-positive bacterium M. nematophilum, which, after its passage through the intestine (Parsons and Cipollo, 2014), adheres to the rectal cuticle and induces a tail-swelling response, the Dar (deformed anal region) phenotype (Hodgkin et al., 2000), as well as changes in intestinal gene expression (O'Rourke et al., 2006). The tail-swelling response has been shown to be a protective response that is associated with prevention of intestinal colonization, severe constipation, and sterility. As the response to M. nematophilum involves the dramatic, readily observed Dar phenotype, this has been used in direct genetic screens to identify mutants with alterations in bacterial colonization of the cuticle and/or intestine (Gravato-Nobre et al., 2005; Yook and Hodgkin, 2007). It was known that activation of the ERK pathway could trigger a Dar phenotype and genetic analysis showed the MPK-1 ERK MAPK pathway to be required for the protective swelling response provoked by adhesion of M. nematophilum. The Ras protein LET-60, the RAF MAPKKK LIN-45, and the MEK MAPKK MEK-2, which function upstream of MPK-1 in vulval development, also activate MPK-1 in the rectal epithelial cells and are required for resistance to M. nematophilum (Nicholas and Hodgkin, 2004; McMullan et al., 2012). Within the rectal epithelium, a conserved Gαq-RhoGEF Trio-Rho signaling pathway (EGL-30, UNC-73, RHO-1) cooperates with the Ras pathway to trigger changes in cell morphology via the RAF-MEK-ERK cassette (McMullan et al., 2012).
The strength of the morphological changes that accompany M. nematophilum infection depends on the developmental stage: L1 and L2 worms exhibit a weaker Dar phenotype than later larval stages, and adults show little or no swelling unless infected at the L4 stage or earlier, perhaps reflecting a loss of the potential for tissue remodeling. Infection of L3 or L4 larvae provokes a strong Dar phenotype visible within 6 h of exposure to M. nematophilum (Powell and Ausubel, 2008). These morphological changes are accompanied by rapid alteration in gene expression, particularly of defense genes in the intestine (O'Rourke et al., 2006). As has been pointed out previously, there is a marked overlap between the genes that are induced upon infection with M. nematophilum and with P. aeruginosa (Irazoqui et al., 2010). Although many P. aeruginosa-induced genes are targets of PMK-1 signaling, relatively few of the commonly up-regulated genes are known targets of PMK-1. Those that are include hpo-6, clec-67, and clec-68. It has yet to be established whether the remaining genes are targets of mpk-1 signaling.
Charles Janeway's original paradigm for activation of innate immunity involves the engagement of a set of germline-encoded “pattern recognition receptors”, which recognize common microbial structural moieties, such as lipopolysaccharide from the cell walls of Gram-negative bacteria (Janeway, 1998). Such a model has been largely validated, for example by the identification of mammalian TLRs as receptors for microbe- or pathogen-derived molecular patterns (MAMPs and PAMPs, respectively) (Janeway and Medzhitov, 2002; Akira et al., 2006).
C. elegans, however, lacks many of the cell surface and intracellular receptors involved in innate immunity in other species (Kurz and Ewbank, 2003). There are, for example, no orthologs of peptidoglycan recognition proteins, or of Gram-negative binding proteins, or of NOD-like receptors. And although the one TLR gene in C. elegans (tol-1) modulates behavioral responses to pathogenic bacteria (Pujol et al., 2001; Pradel et al., 2007), TOL-1 does not appear to function directly in pathogen recognition, instead being required for the terminal differentiation of the CO2-sensing BAG neurons (Brandt and Ringstad, 2015). Further, as stated above, it does not regulate AMP gene expression after infection by D. coniospora (Couillault et al., 2004) or S. aureus (Irazoqui et al., 2010).
What evidence supports the existence of specific pathogen recognition in C. elegans? Although worms exposed to different bacteria have distinct transcriptional profiles (Wong et al., 2007), a critical question is whether such changes represent an initial response contingent upon the recognition of the pathogen, or an effect related to the infection-induced pathology. If the infection with different pathogens progresses through distinct sets of pathological changes, defense mechanisms could appear to be pathogen-specific even in the absence of any microbial recognition mechanism. With such a caveat in mind, the comparison of the gene expression programs of worms exposed to live and dead, virulent and avirulent pathogens (Troemel et al., 2006; Irazoqui et al., 2010), suggests a very complex regulation of host responses (Table 2). Moreover, the expression of certain defense genes appears to be exquisitely sensitive to environmental conditions. For example, leaving worms too long in the standard M9 buffer is sufficient to provoke an upregulation of nlp-29 (N. Pujol and J. Ewbank, personal communication, The Worm Breeder's Gazette). This AMP gene is also induced in worms put under other conditions of relatively mild osmotic stress, for example, if cultured on plates that have become somewhat desiccated. As this was not known at the time, it is probably the reason why nlp-29 was erroneously identified in early studies as being up-regulated by the bacterial pathogen S. marcescens (Wong et al., 2007). Perhaps underscoring this sensitivity to environmental conditions, and at the same time reflecting an overlap in the control of genes in response to pathogens and other stressors (see Section 5), among the list of 263 genes that are induced by starvation (Laing et al., 2012) there are a surprising number of genes categorized as encoding effectors of innate immunity, including abf-5, cnc-4, fipr-22, fipr-23, ilys-2, ilys-3, nlp-28, nlp-29, thn-1, and thn-2, as well as the transcription factor gene zip-2. There is thus a significant overlap between this list of genes and, for example, genes identified as being induced in worms after infection with S. aureus (Irazoqui et al., 2010). This suggests that some pathogens may induce a starvation-like response.
Table 2. Induction of gene expression by live, dead and attenuated P. aeruginosa.
| Sequence Name | Gene Name | Live PA14 | Dead PA14 | gacA mutant |
|---|---|---|---|---|
| F56D6.2 | clec-67 | Y | Y | Y |
| K08D8.5 | Y | Y | Y | |
| F35E12.5 | Y | Y | N | |
| C17H12.8 | Y | Y | N | |
| F53E10.4 | irg-3 | Y | N | Y |
| C49G7.5 | irg-2 | Y | N | Y |
| F01D5.5 | Y | N | Y | |
| F49F1.6 | mul-1 | Y | N | Y |
| F55G11.2 | Y | N | N | |
| C32H11.12 | dod-24 | Y | N | N |
Distinct transcriptional profiles in response to different bacterial pathogens have been interpreted in terms of likely distinct recognition mechanisms, but such data are indirect, and a direct role for specific proteins mediating recognition to pathogens has remained elusive. Motivated by the presence of leucine-rich repeats (LRRs) in TLRs and a number of other classes of innate immune receptors in animals and/or plants, a reverse genetic analysis of worm genes with extracellular LRRs that might function in pathogen defense was performed, resulting in the identification of FSHR-1, homolog of a mammalian hormone receptor. Genetic analysis suggests that FSHR-1 functions in parallel to both p38 MAPK and ZIP-2 (see Section 4.2) pathways in the intestine (Powell et al., 2009); however, a role for FSHR-1 in pathogen recognition has not been demonstrated. Indeed, a very recent study has shown that FSHR-1 is required for resistance of C. elegans to xenobiotics that cause oxidative stress, as well as to infection by diverse pathogens (Miller et al., 2015).
Interestingly, as mentioned above, C. elegans possesses a large repertoire of genes encoding galectins and C-type lectins, as well as F-box proteins (222, 113, and 55 of the A (fbxa), B (fbxb), and C (fbxc) gene classes, respectively). Lectins bind carbohydrates, and F-box proteins, acting as substrate-specific adaptors, mediate ubiquitination of proteins targeted for degradation by the proteasome. These different classes have all been proposed to participate in pathogen recognition (Nicholas and Hodgkin, 2004; Thomas, 2006), and although to date there is no direct experimental evidence to support such a supposition, these gene classes are highly variable in different natural isolates of C. elegans, perhaps reflecting differences in the microbial environments of the strains (Volkers et al., 2013).
There are also a number of potential scavenger receptors (SR), including the SCARF ortholog CED-1, and 6 SCAV proteins homologous to the SR-B family members CD36 and Croquemort. A proposed role for SCAV-1 in microbial recognition (Nicholas and Hodgkin, 2004) was lent support by its apparent contribution to host resistance to Candida albicans and Cryptococcus neoformans in C. elegans; loss of function mutants succumb prematurely to infection (Means et al., 2009). ced-1 mutants also display a similar increased susceptibility to both intestinal fungal pathogens, but in this case the well-established role of ced-1 in apoptotic cell corpse engulfment makes interpretation of the results more complex. Indeed, whether these C. elegans proteins actually recognize yeast cell wall β-glucans and thereby trigger downstream effector gene expression has not been formally demonstrated. It is noteworthy that knocking down scav-4 increases susceptibility to the nematocidal toxin Cry5B (Kao et al., 2011), arguing in favor of an indirect role in host defense.
Janeway's original model for activation of innate immunity that involves the engagement of MAMP and PAMP receptors (Janeway, 1998) was refined by the inclusion of molecules associated with damage and cellular injury (DAMPs, for “damage-associated molecular patterns”, or “danger signals”) as triggers of the innate immune response (Matzinger, 2002; Seong and Matzinger, 2004). Whereas direct evidence for pathogen recognition activating innate immunity in C. elegans has been lacking, recent studies point to the activation of host defense pathways by processes affected by cellular damage in C. elegans.
As detailed above in Section 3, genetic studies implicate a pivotal role for the PMK-1 p38 MAPK pathway in innate immunity in the intestine and epidermis. Two lines of study have demonstrated that PMK-1 p38 MAPK is activated by cellular damage and injury in C. elegans. First, direct wounding of the epidermis with a laser or a needle can induce the same PMK-1-dependent antifungal gene expression as is observed during fungal infection of the epidermis of C. elegans (Pujol et al., 2008a; Pujol et al., 2008b). Second, the lethal action of pore-forming toxins expressed by bacteria has been shown to activate PMK-1 signaling in the intestine of C. elegans, and the PMK-1 pathway contributes to resistance to the action of pore-forming toxins (Huffman et al., 2004; Bischof et al., 2008). Pore-forming toxins form large diameter channels in the membrane that cause loss of cellular contents and of homeostasis. A JNK-like MAPK, KGB-1, has additionally been shown to be required for resistance to pore-forming toxins (Huffman et al., 2004), to P. aeruginosa infection (Kim et al., 2004), and to heavy metal toxicity (Mizuno et al., 2004), implicating this MAPK also in response and repair mechanisms to diverse insults.
Studies of the role of calcium, however, point to some specificity in the response to damage and infection. Intracellular calcium increases upon wounding in C. elegans and, through activation of a Gαq-PLCβ (EGL-30-EGL-8) pathway, this is essential for tissue repair. While calcium-dependent signaling mechanisms have been shown to function upstream of ASK1/NSY-1 activation in different contexts (e.g., Troemel et al., 1999; Sagasti et al., 2001), calcium release does not seem to be required for the induction of AMP gene expression (Xu and Chisholm, 2011).
Nevertheless, the involvement of common MAPK pathways, not only in the response to pathogenic bacteria but also to wounding and exposure to pore-forming toxins, supports the possibility of a common mechanism activating innate immunity that involves the sensing of tissue damage. The study of the regulation of antimicrobial peptides in the epidermis has recently given insights into two such mechanisms. In one of these, it appears that compromising the integrity of the epidermis, either by physical injury or in mutants such as dpy-9 and dpy-10, leads to an increase in the concentration of hydrophenyllactic acid (HPLA). This tyrosine-derivative then activates its cognate receptor DCAR-1, which acts upstream of gpa-12 and the tir-1/p38 MAPK cascade to switch on AMP gene expression via sta-2 (Zugasti et al., 2014).
Antifungal peptide expression in the epidermis also increases in response to severe tissue damage. This was reported to proceed through a STA-2-dependent mechanism, apparently mediated by the mechanical sensing of membrane damage through the hemidesmosomal protein MUP-4, independently of PMK-1 (Zhang et al., 2015). STA-2 was shown to be physically associated with hemidesmosomes that attach the hyp7 plasma membrane to the cuticle. It was proposed that this interaction could be disrupted when the epidermis is damaged, liberating STA-2, which would then be free to translocate to the nucleus and activate the expression of AMP gene expression. It should be noted, however, that there was no demonstration of the expected increase in nuclear STA-2. On the other hand, hemidesmosome disassembly in primary human epidermal keratinocytes also induces AMP gene expression, arguing in favor of a conserved damage-sensing defense mechanism (Zhang et al., 2015).
Changes in the translational capacity of the cell can trigger an evolutionarily ancient stress response present in bacteria, yeast, and mammals (Iordanov et al., 1997; Argüello et al., 2014). In eukaryotic cells, treatment with protein synthesis inhibitors switches on stress-activated MAPK signaling (Claudio et al., 2013). The activation of MAPK signaling (and specifically PMK-1 p38 MAPK) by translational inhibition, including by bacterial toxins, has recently been shown to be conserved in C. elegans (Dunbar et al., 2012; McEwan et al., 2012; Chou et al., 2013). Thus ribosomal stress and translational inhibition can activate the same signaling cassette that is activated by infection with pathogenic bacteria, and have been speculated to serve as a conserved trigger for the activation of innate immunity (Mohr and Sonenberg, 2012).
Infection of C. elegans by P. aeruginosa PA14 also provokes a response that is PMK-1-independent and that relies instead on ZIP-2, a bZIP transcription factor that does not have an identifiable ortholog in higher animals. ZIP-2 was found to function in the intestine to regulate the expression of many genes induced by infection with P. aeruginosa and to be required for pathogen resistance (Estes et al., 2010). ZIP-2 regulates the expression of many genes, including the Infection Response Genes irg-1 and irg-2 that are induced by P. aeruginosa infection. ZIP-2-regulated genes are also upregulated by inhibitors of translation (Dunbar et al., 2012; McEwan et al., 2012). Further, the expression of ZIP-2 itself was observed to increase following PA14 infection or pharmacological inhibition of protein translation. The translational control of ZIP-2 expression was shown to be mediated by a regulatory element in the 5’UTR of the zip-2 mRNA (Dunbar et al., 2012). The PA14 gacA mutant, in which expression of the translation-blocking toxin ToxA is diminished, does not cause the up-regulation of the two irg genes (Troemel et al., 2006; Estes et al., 2010). While the contribution of specific microbial toxins to the activation of host immune responses in other infection models remains to be determined, these data suggest that interfering with cellular physiology can activate signal transduction pathways that are induced by pathogen infection.
Cellular defense mechanisms can also be triggered by abiotic stress caused, for example, by alterations in osmolarity or temperature, or by exposure to heavy metals. These stressors can provoke the accumulation of unfolded proteins and reactive oxygen species. Although these stresses have generally been considered distinct from those induced by pathogens, there is evidence to suggest that much of the “abiotic” stress experienced by C. elegans in its natural environment comes from contact with microbes, through their production of toxins, or as a consequence of the host response to infection. Moreover, as first shown by studies of the response to nematocidal pore-forming toxins, which act by directly damaging cellular membranes (Huffman et al., 2004), the mechanisms mediating stress responses intersect with pathways regulating innate immunity. The juxtaposition of innate immunity and stress responses has been particularly evident in recent studies that link the disruption of basic cellular mechanisms, such as translation (see Section 4.2) or mitochondrial homeostasis, to anti-microbial defenses (Melo and Ruvkun, 2012; Liu et al., 2014; Pellegrino et al., 2014; Kirienko et al., 2015). In this section, we consider further the interplay between stress response pathways and innate immunity, reviewing the evidence for the involvement of distinct stress response pathways during pathogen infection and considering the hypotheses that have been proposed regarding innate immune activation and stress responses.
Starting from the observation that a GATA motif is found in the promoter sequences of a disproportionately high fraction of genes that are differentially regulated following PA14 infection, Tan and colleagues demonstrated that the GATA factor ELT-2 is required for normal resistance to P. aeruginosa (Shapira et al., 2006). Because elt-2 is an essential gene in intestinal morphogenesis (Fukushige et al., 1998), RNAi-based analysis was used to implicate ELT-2 in the regulation of the innate immune response, including data that elt-2(RNAi) significantly reduced basal and infection-induced expression of certain candidate genes, which are therefore likely to be direct targets of ELT-2 (McGhee et al., 2009). The role of elt-2 was relatively specific: when knocked down there was no alteration in resistance to oxidative stress caused by paraquat, 37°C heat stress, or exposure to cadmium (Shapira et al., 2006). On the other hand, elt-2 does play an important role in regulating organismal resistance to osmotic stress, in part through the transcriptional control in the intestine of gpdh-1 (Rohlfing et al., 2010), which encodes the rate-limiting enzyme for the production of the osmoprotectant glycerol. Beyond this, there is a substantial overlap between the intestinal genes up-regulated by PA14 infection and as part of the response to osmotic stress (Rohlfing et al., 2010). Similarly, the epidermal-specific factor GATA factor ELT-3 (Tonsaker et al., 2012) is required for the basal and infection-induced expression of an nlp-29p::GFP reporter, as well as for the induction upon osmotic stress of gpdh-1 expression specifically in the epidermis (Pujol et al., 2008b). Again, there is a substantial overlap between the genes induced upon D. coniospora infection and by osmotic stress. Infection and osmotic stress may provoke an overlapping spectrum of perturbations in cellular physiology, such as an inhibition of protein synthesis (Choe and Strange, 2008; Burkewitz et al., 2012; Lee and Strange, 2012), that have as a consequence a shared transcriptional response.
It is interesting to note that ELT-2 and ELT-3 act in concert with DAF-16 (see Section 5.2) in the intestine and epidermis, respectively, to modulate gene expression under normal culture conditions (Zhang et al., 2013). The potential importance of GATA factors in host defense is underscored by the finding that ELT-2 is degraded by the host ubiquitin–proteasome system following infection with Burkholderia pseudomallei, in a process dependent upon this bacterial pathogen's type III secretion system (Lee et al., 2013).
As discussed in a recent review (Kim, 2013), there is interplay between innate immunity and longevity. One central player in this is daf-2, an insulin receptor-like gene. Mutations that reduce daf-2 function confer dramatic enhancement in resistance to pathogenic bacteria that is dependent on the conserved Forkhead transcription factor DAF-16 (Garsin et al., 2003). The DAF-2-DAF-16 signaling pathway was initially characterized for its role in the regulation of entry into dauer diapause and because daf-2 mutant worms exhibit a dramatic DAF-16-dependent extension in lifespan when cultured on E. coli OP50 (Kenyon et al., 1993; Kimura et al., 1997). DAF-16 has also been shown to mediate resistance to heat, oxidative stress, and several other abiotic stressors (e.g., Barsyte et al., 2001; Henderson and Johnson, 2001; Ookuma et al., 2003; Hertweck et al., 2004; Lamitina and Strange, 2004; Liang et al., 2006). A number of signaling components have been identified that function in conjunction with or in parallel to DAF-16 downstream of DAF-2 to affect stress resistance and longevity, including some, such as SKN-1, a Cap'n'collar (CnC) family transcription factor homologous to mammalian Nrf2, and the heat shock factor HSF-1, which have established roles in stress response signaling (Hsu et al., 2003; Viswanathan et al., 2005; Wolff et al., 2006; Tullet et al., 2008; Wang et al., 2010; Seo et al., 2013; Tullet et al., 2014).
In view of the enhanced resistance of the daf-2 mutant to a diverse range of stressors, as well as its extended lifespan, the pathogen resistance of this mutant is perhaps not that surprising. And yet, this observation raises the question of how much specificity exists in the mechanisms that confer resistance to abiotic stressors or pathogens and that extend lifespan. That is, how much does stress resistance contribute to pathogen resistance? Indeed, in one of the first studies in the field, Mahajan-Miklos et al. demonstrated a hormetic effect (positive results from exposure to low doses of an agent that is otherwise toxic or lethal at higher doses) of developmental stress on adult pathogen resistance (Mahajan-Miklos et al., 1999). A further example of hormesis derives from a study of the effect of growing worms on pathogenic bacteria. It was found that exposure to particular pathogens during development, while not affecting larval survival, increased the lifespan of adult worms exposed to the same or a different pathogen. This increased resistance did not result from an enhanced capacity to kill bacteria. Rather, it was associated with an increase of chaperone gene expression and higher resistance to heat shock. It may therefore be due to an increased tolerance to the damage inflicted by pathogenic bacteria (see Section 5.3). Interestingly, this effect was dependent upon the DBL-1, DAF-2/DAF-16, and PMK-1 p38 MAP kinase pathways (Leroy et al., 2012).
Transcriptional profiling suggests that at least in the case of DAF-16, the contributions of stress response genes and innate immune effectors overlap to promote pathogen resistance. Genome-wide analyses have suggested that DAF-16 regulates many genes with putative antimicrobial activities, in addition to genes involved in stress resistance and detoxification (McElwee et al., 2003; Murphy et al., 2003; Oh et al., 2006; Zhang et al., 2013; reviewed in Landis and Murphy, 2010). The pathogen resistance and lifespan of daf-2 mutants is strongly dependent on the activity of the PMK-1 pathway, but these data are consistent with the PMK-1 pathway functioning either downstream of or parallel to insulin signaling to promote pathogen resistance (Troemel et al., 2006). Additional studies have indicated further interplay between DAF-16 and cellular and organismal stress-activated signaling (Kondo et al., 2005; Sem et al., 2012; Tao et al., 2013).
If DAF-16 functions to respond to pathogen infection, one might anticipate the induction of DAF-16 activity in response to pathogen infection. Surprisingly, studies suggest the opposite: that pathogenic P. aeruginosa infection may result in the suppression of DAF-16 activity (Evans et al., 2008; Kawli and Tan, 2008). Does this represent the pathogen targeting a host innate immune signaling pathway to promote pathogenesis, or does this observation suggest that the role of DAF-16 in mediating host survival is secondary to the initial innate immune and stress responses induced during infection? Addressing such questions will require further research.
The accumulation of unfolded proteins in the cell induces organelle-specific responses. The cellular heat shock response involves the activation of conserved stress signaling mechanisms in the cytosol that converge on HSF-1. This transcription factor was previously noted to be required for the longevity and heat resistance of daf-2 mutants, and a role for HSF-1 in pathogen resistance has also been demonstrated (Singh and Aballay, 2006, Singh and Aballay, 2009).
The Unfolded Protein Response (UPR) maintains endoplasmic reticulum (ER) homeostasis. Genetic analysis of UPR genes revealed an essential role for the conserved transcriptional regulator XBP-1 in survival on pathogenic bacteria of C. elegans specifically during larval development (Richardson et al., 2010). Infection with P. aeruginosa (Richardson et al., 2010) or intoxication with pore-forming toxins (Bischof et al., 2008) induced the activation of XBP-1 in response to ER stress. Surprisingly, mutation in pmk-1 suppressed the requirement for XBP-1 during development in the presence of P. aeruginosa. This apparently paradoxical finding was proposed to result from a decrease in immune effector synthesis lessening the secretory load on the ER and thereby increasing host fitness (or resilience) in the absence of XBP-1 function, so permitting survival (Ewbank and Pujol, 2010; Richardson et al., 2010; Richardson et al., 2011). These data point to an ancient, essential role for XBP-1 and the UPR in maintaining ER homeostasis in response to the endogenous ER stress generated by the innate immune response to infection by pathogenic bacteria. Mechanisms that enable a host to survive pathogen infection without directly diminishing pathogen load have been termed “tolerance” (Medzhitov et al., 2012), and the finding that the UPR enables a full innate immune response provides a striking illustration of such tolerance.
In addition to the conserved xbp-1-dependent UPR, a set of genes proposed to function in a “non-canonical UPR,” composed of the abu (Activated in Blocked Unfolded protein response) family genes, has been suggested to play an important role in host defense (Haskins et al., 2008; Sun et al., 2011). A recent study of abu gene expression and function, however, casts doubt on their presumed role in ER homeostasis and host defense (George-Raizen et al., 2014). The abu genes were originally identified as being upregulated in xbp-1 mutant animals (Urano et al., 2002). Recently, the abu genes were shown to exhibit several hundred-fold changes in expression during the molt. Because these genes are subject to such marked developmental regulation, comparisons of their expression between populations can only be made if extreme care is taken to ensure that they are precisely synchronized (George-Raizen et al., 2014). Indeed, with careful staging of animal populations, the previously reported changes in abu gene expression due to mutations, pathogenic bacteria, and ER stressors were not observed. Furthermore, the abu genes have been shown to encode components of the pharyngeal cuticle (George-Raizen et al., 2014), so that reduction of abu gene function may cause the observed effects on susceptibility to pathogenic bacteria through alterations in pharyngeal morphology and efficiency. It should be further noted that feeding-defective mutants leave and avoid non-pathogenic bacteria (Olofsson, 2014). These studies serve as a cautionary tale on the limits of using gene expression analysis to infer specific function in host defense.
Whereas the study of XBP-1-dependent signaling suggests that the UPR is induced by activation of innate immunity, recent studies suggest that proteotoxic stress in the mitochondria can function to induce the intestinal innate immune response. Exposure of worms to P. aeruginosa and to a range of commonly encountered environmental bacteria was shown to provoke the mitochondrial UPR, which in turn activates signaling components of the innate immune response (Liu et al., 2014; Pellegrino et al., 2014). These studies raise the intriguing possibility that cellular innate immune responses may be activated by the action of microbe-derived toxins that perturb mitochondrial protein folding homeostasis.
Autophagy is a cellular homeostatic mechanism that has been shown to be important under cellular stress conditions, assisting in the maintenance of membrane homeostasis. It can help clear damaged organelles, such as mitochondria, and confers resistance to siderophore-mediated killing during P. aeruginosa infection (Kirienko et al., 2015). It has also been implicated in immunity to intracellular pathogens. In C. elegans, autophagy contributes to resistance to Salmonella infection by mechanisms that are still to be elucidated (Curt et al., 2014). A recent study also compared genes induced by S. aureus infection with those regulated by the autophagy transcriptional regulator HLH-30 (the nematode homolog of vertebrate TFEB) and found a large degree of overlap (Visvikis et al., 2014). Starvation is a known inducer of autophagy, and as S. aureus is a poor food source for C. elegans, some proportion of the observed overlap in transcriptional changes induced by S. aureus may be due to the indirect effects of S. aureus on host metabolism. Nevertheless, in L4 larvae and young adults, HLH-30/TFEB was shown to be translocated to the nucleus after only 30 minutes exposure to S. aureus and to be required for host defense against S. aureus, suggestive of a role for autophagy in C. elegans host defense (Visvikis et al., 2014).
Reactive oxygen species (ROS) comprise an important component of the antimicrobial response of immune cells. The genetic analysis of Drosophila antimicrobial defenses has suggested an important protective role for the generation of ROS in the intestinal lumen (Ha et al., 2005). In host defense, ROS are generated by NADPH dual oxidase (DUOX) enzymes. It was recently shown that bacterially-derived uracil can activate intestinal DUOX in Drosophila. Preliminary results indicate that this is also the case in worms (Lee et al., 2013). Earlier genetic studies in C. elegans showed that reducing expression of the nematode DUOX BLI-3 increases susceptibility to Enterococcus faecalis infection, as well as causing a detectable decrease in H2O2 production (Chavez et al., 2009). This led to the suggestion that BLI-3 might generate H2O2 and ROS that could be released into the intestinal lumen during infection, thereby contributing to pathogen killing (Chavez et al., 2009). An accompanying role for the peroxidase SKPO-1 in this process has also been proposed (Tiller and Garsin, 2014). Although the expression of bli-3 and skpo-1 in the epidermis, and their essential role in the formation of collagen crosslinks in the cuticle (Edens et al., 2001; Moribe et al., 2012; Tiller and Garsin, 2014), complicates the interpretation of pathogen susceptibility data, recent work has confirmed that BLI-3 is expressed in the intestine (van der Hoeven et al., 2015).
ROS can be toxic and can compromise host survival. The release of ROS during innate immune activation thus necessitates a host response to oxidative stress to limit toxicity. Oxidative stress induces activation of PMK-1, which activates the expression of detoxification genes controlled by SKN-1 (Inoue et al., 2005) and the RUNX transcription factor homolog RNT-1 (Lee et al., 2012), thus conferring protection against oxidative stress (Inoue et al., 2005). Consistent with the idea that a response to ROS may help protect the host during infection (Miller et al., 2015), exposure to P. aeruginosa can lead to activation and nuclear translocation of SKN-1 in the intestine (Papp et al., 2012). There are, however, conflicting results regarding whether mutation of skn-1, or RNAi of skn-1, confers enhanced susceptibility to killing by pathogenic bacteria (Kawli et al., 2010; Shivers et al., 2010; Hoeven et al., 2011).
As outlined in Section 3 and Section 4.2, MAPK signaling is important both for pathogen resistance and for abiotic and more generic stress responses, including chronic osmotic stress (Solomon et al., 2004; Wheeler and Thomas, 2006). Some specificity in PMK-1 p38 MAPK signaling is provided by distinct transcription factor targets, which regulate specific outputs. Thus, for example, activation of SKN-1 and RNT-1 confer resistance to oxidative stress, and ATF-7 contributes to protection from pathogenic bacteria. Whether PMK-1 activates ATF-7 in response to oxidative stress in the absence of infection remains unclear. ROS mediate the activation of the ASK1-p38 MAPK pathway downstream of TLR activation in mice (Matsuzawa et al., 2005). Thus they may have a conserved role in innate immune signaling. The conserved Mediator subunit MDT-15/MED15 was shown to be required for the induction of stress-responsive and pathogen-induced genes regulated by the PMK-1 pathway, suggesting a role for MDT-15/MED15 in the coordination of detoxification activities and innate immunity (Pukkila-Worley et al., 2014).
The involvement of PMK-1 in both innate immunity and responses to oxidative stress suggests that the activation of specific transcription factors by PMK-1 represents an important node of integrating information regarding diverse stressors. Additional cross-regulation with stress responses occurs at the level of distinct stress-activated MAPK pathways. MAPK phosphatases have regulatory roles in modulating the activities of MAPK signaling pathways. The MAPK phosphatase VHP-1 regulates both PMK-1 and the JNK-like MAPK KGB-1, which has a role in resistance to heavy metal stress (Mizuno et al., 2004). Thus, VHP-1 may represent an important point of integrating signal transduction through stress-activated and innate immune responses.
Interestingly, kgb-1 mutants exhibit enhanced susceptibility to heavy metals, to the toxin tunicamycin, which causes ER stress, and to killing by P. aeruginosa (Kim et al., 2004; Mizuno et al., 2004). More recently, kgb-1 was also shown to be required for the ER UPR provoked by paraquat (Runkel et al., 2013). The MAPKK MEK-1 activates KGB-1. Prior studies suggested the possible cross-regulation of PMK-1 by MEK-1, but the identification of a tightly linked partial loss-of-function mutation in sek-1(qd127) in the strain carrying the mek-1(ks54) mutant allele (K. Reddy and D. Kim, unpublished data) suggests that the diminished PMK-1 activation observed in this mutant strain may actually be due to diminished SEK-1 activity. If MEK-1 has a role in mediating pathogen resistance, it is likely to be solely through the activation of KGB-1.
In the context of stress resistance, the bZIP transcription factor FOS-1 has shown to be a target of KGB-1 phosphorylation. Unphosphorylated FOS-1 recruits the histone deacetylase HDA-1 to the promoters of stress-inducible genes, where it acts as a transcriptional repressor. KGB-1's phosphorylation of FOS-1 prevents its dimerization, thereby impairing promoter binding of the HDA-1/FOS-1 complex, leading to derepression of target gene expression (Hattori et al., 2013). It will be interesting to see whether the contribution of kgb-1 to resistance to pore forming toxins is mediated by the same mechanism.
Establishing whether genes that contribute to resistance to pathogenic bacteria do so through changes in innate immunity, stress signaling, or perhaps affect another aspect of physiology such as behavior can be challenging. Many of the mechanisms that have been discussed above are cell-autonomous. Host defense responses to infection, however, involve broad physiological changes and multicellular organisms need to integrate the immune responses with other aspects of physiology, and this can necessitate communication between tissues. In C. elegans, for example, intestinal infection with Salmonella provokes an increase in apoptosis in the germline (Aballay and Ausubel, 2001). Conversely, loss of DNase II function or DNA damage in the nematode germline is sufficient to induce the ERK MAPK pathway, the expression of innate immune effector genes, and a systemic resistance to stress via activation of the ubiquitin-proteasome system (UPS) in somatic tissues (Ermolaeva et al., 2013; Yu et al., 2015). As a last example, germline ablation can lead to increased resistance to P. aeruginosa infection, via a DAF-16 independent pathway (Alper et al., 2009).
There is considerable interest in the question of how the nervous system might influence the immune system. This has been investigated in a number of organisms, but C. elegans, despite its lack of dedicated immune cells, represents perhaps the most tractable host organism to explore the basic molecular mechanisms underlying such interactions because of the detailed understanding of the anatomy and physiology of the 302-cell nervous system. Even in C. elegans, however, understanding such links requires close scrutiny, since neuronal control of avoidance behavior, as well as other aspects of physiology such as egg-laying, may affect resistance to killing by pathogens without directly affecting the innate immune response (Table 3). Further, there is evidence for coordinated regulation in different tissues by common signaling cassettes. Specific examples of the modulation of defense gene expression involving neurons are given below.
Table 3. Neuronally-expressed genes proposed to regulate innate immunity
| Gene | Protein Homolog | Evidencea | Caveatb | References | |||
|---|---|---|---|---|---|---|---|
| Tr | Hom | GoF | LoF | ||||
| dbl-1 | TGF-ß | X | X | X | X | Controls morphology and polyploidy | (Mochii et al., 1999; Mallo et al., 2002; Zugasti and Ewbank, 2009; Roberts et al., 2010) |
| ins-7 | insulin | X | X | X | X | Controls DAF-16 activity | (Evans et al., 2008; Kawli and Tan, 2008; Engelmann et al., 2011) |
| npr-1 | neuropeptide Y receptor | X | X | Relatively small changes of effector gene expression in npr-1 mutants; influences development and physiology | (Styer et al., 2008; Reddy et al., 2009; Andersen et al., 2014) | ||
| octr-1 | 5-hydroxytryptamine receptor 1B | X | X | Transcriptional targets include genes required for pharynx function | (Sun et al., 2011; George-Raizen et al., 2014) | ||
|
aiv.: Tr: altered transcription of known or putative immune effector genes in mutant (gain- or loss-of-function). change upon infection; Hom: homolog(s) in other species with demonstrated immune regulatory function; Gof: genetic gain-of-function (e.g., over-expression leads to protection); Lof: genetic loss-of-function (e.g., RNAi leads to susceptibility). bFactors or additional roles that could potentially explain changes in resistance and/or the expression of innate immune gene expression. |
|||||||
INS-7. The insulin-like peptide gene ins-7 is expressed in the nerve ring, amphid sensory, labia, ventral cord, and tail neurons. Release of INS-7 from dense core vesicles in neurons has been shown to inhibit DAF-16 signaling in the intestine and thereby negatively-regulate pathogen resistance (Kawli and Tan, 2008). Following infection with wild-type P. aeruginosa, but not in mutants with attenuated virulence, ins-7 expression is induced leading to the down-regulation of putative immune defense genes including lys-7 and thn-2. These observations led to the suggestion that P. aeruginosa has developed a specific mechanism to subvert immune defense via modulation of a host neuro-immune regulatory pathway (Evans et al., 2008).
DBL-1. In another example, the TGFβ ligand DBL-1 has been shown to function in the non-cell-autonomous control of defense gene expression in response to the fungus D. coniospora. As mentioned above, fungal infection of the epidermis induces not only the nlp AMP genes, but also provokes the PMK-1-independent induction of cnc genes. In a dbl-1 mutant the induction of several cnc genes, (cnc-1, 2, 4, 5, and 11), is greatly diminished, while the induction of the nlp AMP genes is unaffected. Expression of dbl-1 under the control of the rab-3 pan-neuronal promoter is sufficient to restore cnc-2 gene induction in the epidermis upon infection. The canonical dbl-1 pathway (see Signaling in the immune response, Figure 1) influences many physiological and developmental traits, including intestinal and epidermal polyploidy and body length. Interestingly, in the context of cnc gene regulation, sma-2 and sma-4 are dispensable, suggesting that the pre-existing dbl-1 pathway was co-opted and modified to contribute to immune defense (http://www.wikipathways.org/index.php/Pathway:WP2233). In contrast to the regulation of the DAF-2/DAF-16 pathway by INS-7, there is no evidence to suggest that infection alters the secretion of DBL-1 (Zugasti and Ewbank, 2009). The current model is that infection triggers a maturation and activation of a pre-existing pool of the precursor form of DBL-1.
|
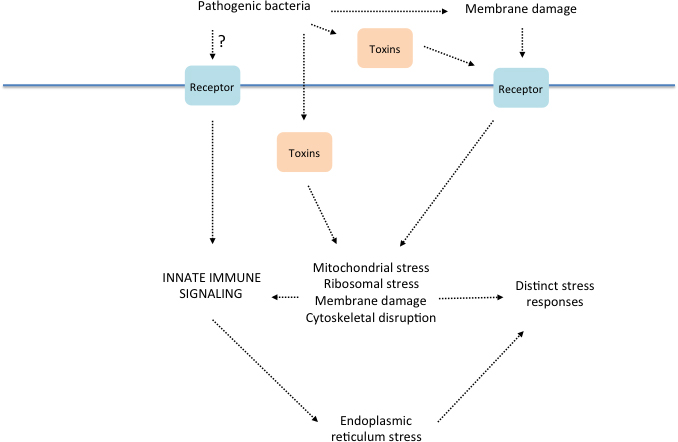
Figure 3. Relationships between the innate immune response to bacterial infection and cellular stress mechanisms. Speculative model for diverse triggers of defense gene expression in response to infection, their consequences and their inter-relationships. This figure is a simplified adaptation of a more complete model from a recent concise but comprehensive review (Cohen and Troemel, 2015).
There are indications that dbl-1 may also influence the response to intestinal pathogens. A number of the genes that are induced by the intestinal bacterial pathogen S. marcescens, including F55G11.4, lys-1, 7, and 8, are known targets of dbl-1 signaling (Mochii et al., 1999; Mallo et al., 2002; Alper et al., 2007; Roberts et al., 2010), and dbl-1 mutants have a reduced resistance to infection (Mallo et al., 2002). Although the details of the underlying regulatory mechanisms have yet to be elucidated, they may be distinct from those that govern expression of AMP genes in the epidermis, as the expression of some of the targets (such as lys-8) is dependent on sma-2 (Mochii et al., 1999).
NPR-1. Compared to wild isolates of C. elegans, the standard laboratory strain N2 carries a mutation (F215V) in the npr-1 gene (de Bono and Bargmann, 1998; McGrath et al., 2009), which encodes a G protein-coupled neuropeptide receptor. This lab-derived polymorphism increases NPR-1 activity, giving rise to a number of distinct developmental and behavioral changes (Andersen et al., 2014). N2 animals are more resistant to killing by P. aeruginosa than the CB4856 wild isolate from Hawaii, which carries the natural 215F allele of npr-1, and npr-1 loss-of-function mutants in the N2 background are more susceptible than wild-type N2 animals (Styer et al., 2008; Reddy et al., 2009). This difference in pathogen susceptibility was attributed to differential regulation of the innate immune system through neuronal NPR-1 signaling (Styer et al., 2008), but differences in behavioral avoidance of the lawn of pathogenic bacteria between the N2 and CB4856 strains were found to be sufficient to account for difference in pathogen susceptibility (Reddy et al., 2009). The inconsistences between the published studies on npr-1 have been discussed elsewhere (Kawli et al., 2010a).
OCTR-1. Mutations in the octr-1 gene encoding a C. elegans octopamine receptor, expressed in a small subset of head and tail neurons, have been shown to confer increased resistance to infection (Sun et al., 2011). The proposed mode of action of OCTR-1 is via negative regulation of the non-canonical UPR involving the abu class of genes. As mentioned above, these genes appear to be involved in pharyngeal cuticle and grinder structural integrity. The reported differential expression of abu genes in octr-1 mutants moreover points to differences in developmental rate that could also contribute to the observed differences in pathogen resistance.
Serotonin. Infection with pathogenic P. aeruginosa has been shown to induce the expression of tph-1, which encodes the enzyme necessary for the rate-limiting step in serotonin biosynthesis in the ADF sensory neurons. Serotonin is required for the aversive learning behavior exhibited by C. elegans in response to infection (Zhang et al., 2005). The site of action of serotonin has not been defined, but interestingly, serotonin signaling in chemosensory neurons appears to also be involved in the distinct response to M. nematophilum, modulating the activity of G-protein signaling in the epithelial cells of C. elegans (Anderson et al., 2013).
These examples demonstrate that further study of the integration of innate immune signaling and different aspects of physiology at the molecular and organismal level is clearly merited.
In addition to the transcriptional regulation of innate immune effector gene expression discussed above, several studies have suggested a role for post-transcriptional regulation. It is important to reiterate that pathogen resistance can be affected for non-specific reason, such as alterations of developmental timing, so that each study must be viewed critically. That said, Kudlow et al. (2012) reported that inactivation of the miRNA-induced silencing complex (miRISC) and mutation of certain miRNA genes provokes overexpression of pathogen-responsive genes under normal culture conditions and enhances worm survival on P. aeruginosa, suggesting that miRNAs are an important part of the regulatory circuit that controls the capacity of C. elegans to resist infection.
One level of control is apparently provided by the regulation of the let-7 family of miRNAs by the nuclear hormone receptor gene daf-12 (Hammell et al., 2009), which integrates environmental signals and developmental timing. Indeed, the developmental timing defects of certain let-7-Fam miRNA mutants are enhanced upon infection with P. aeruginosa (Ren and Ambros, 2015). Loss of daf-12 results in an increase in resistance to P. aeruginosa that is dependent upon nsy-1 and pmk-1. In the authors’ model, daf-12 regulates let-7 family miRNAs, these target skn-1, and SKN-1, activated by the p38 MAPK pathway, controls the expression of genes that contribute to host resistance (Liu et al., 2013; Ren and Ambros, 2015).
Moreover, recently infection of C. elegans by P. aeruginosa was also shown to provoke a PMK-1 p38 MAPK-dependent induction of mir-233 expression. This miRNA governs the expression of sca-1, which encodes a sarco/endoplasmic reticulum Ca2+-ATPase. The consequent reduction in SCA-1 levels was proposed to activate a protective UPR in the worm intestine (Dai et al., 2015).
Endogenous siRNAs have also been suggested to be important in regulating defense genes, in part via PDK-1 which controls both SKN-1 and DAF-16 activity (Mansisidor et al., 2011). With the advent of “social RNA” and the first demonstration of a microbe hijacking host RNAi pathways to suppress host immunity (Sarkies and Miska, 2013; Weiberg et al., 2013), this is an area ripe for new discoveries.
Our understanding of innate immune signaling has progressed substantially since the previous edition of this chapter (Signaling in the immune response). It has become even clearer that innate immunity cannot be analyzed in isolation. There are multiple layers of crosstalk between different developmental and physiological processes that impact the capacity of the host to fight infection. This applies both to the mechanisms that target infectious microbes as well as those that enable the host to resist the damage caused directly (or indirectly) by the pathogen. In such a context, studies that rely on differential gene expression during infection with different microbial pathogens or experimental conditions need to be interpreted with caution, especially since as a category, defense genes have a very dynamic developmental regulation (Capra et al., 2008). Nevertheless, what is emerging is a picture of a patchwork of defenses, both generic and pathogen-specific, that are fully integrated in host physiology. There are many areas that remain to be fully explored—those related to pathogen recognition, the cell biology of the host response, microbial subversion of host defenses, evolutionary questions on the population and species level, and the mechanisms determining resource allocation—to name just a few. This is an exciting time to tackle these subjects.
We thank David Raizen and Niels Ringstad for communicating results before publication, Iva Greenwald for her encouragement, and Nathalie Pujol for constructive comments. Work in the Ewbank lab is supported by institutional funding from INSERM, CNRS and AMU, and program grants from the ANR (ANR-12-BSV3-0001-01, ANR-11-LABX-0054 (Investissements d'Avenir–Labex INFORM), and ANR-11-IDEX-0001-02 (Investissements d'Avenir–A*MIDEX)). Work in the Kim lab is supported by grants from the NIH (GM084477 and AT008764).
Abad, P., Gouzy, J., Aury, J.-M., Castagnone-Sereno, P., Danchin, E.G.J., Deleury, E., Perfus-Barbeoch, L., Anthouard, V., Artiguenave, F., Blok, V.C., et al. (2008). Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 26, 909-915. Abstract Article
Aballay, A., and Ausubel, F.M. (2001). Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc. Natl. Acad. Sci. U. S. A. 98, 2735-2739. Abstract Article
Aballay, A., Drenkard, E., Hilbun, L.R., and Ausubel, F.M. (2003). Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 13, 47-52. Abstract Article
Ahren, D., Tholander, M., Fekete, C., Rajashekar, B., Friman, E., Johansson, T., and Tunlid, A. (2005). Comparison of gene expression in trap cells and vegetative hyphae of the nematophagous fungus Monacrosporium haptotylum. Microbiology 151, 789-803. Abstract Article
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783-801. Abstract Article
Alegado, R.A., Campbell, M.C., Chen, W.C., Slutz, S.S., and Tan, M.W. (2003). Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell. Microbiol. 5, 435-444. Abstract Article
Alegado, R.A., and Tan, M.W. (2008). Resistance to antimicrobial peptides contributes to persistence of Salmonella typhimurium in the C. elegans intestine. Cell. Microbiol. 10, 1259-1273. Abstract Article
Alper, S., McBride, S.J., Lackford, B., Freedman, J.H., and Schwartz, D.A. (2007). Specificity and complexity of the Caenorhabditis elegans innate immune response. Mol. Cell. Biol. 27, 5544-5553. Abstract Article
Alper, S., McElwee, M.K., Apfeld, J., Lackford, B., Freedman, J.H., and Schwartz, D.A. (2009). The C. elegans germline regulates distinct signaling pathways to control lifespan and innate immunity. J. Biol. Chem. 285, 1822-1828. Abstract Article
Andersen, E.C., Bloom, J.S., Gerke, J.P., and Kruglyak, L. (2014). A variant in the neuropeptide receptor npr-1 is a major determinant of Caenorhabditis elegans growth and physiology. PLoS Genet. 10, e1004156. Abstract Article
Andersen, E.C., Gerke, J.P., Shapiro, J.A., Crissman, J.R., Ghosh, R., Bloom, J.S., Félix, M.A., and Kruglyak, L. (2012). Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat. Genet. 44, 285-290. Abstract Article
Anderson, A., Laurenson-Schafer, H., Partridge, F.A., Hodgkin, J., and McMullan, R. (2013). Serotonergic chemosensory neurons modify the C. elegans immune response by regulating G-protein signaling in epithelial cells. PLoS Pathog. 9, e1003787. Abstract Article
Argüello, R.J., Rodrigues, C.R., Gatti, E., and Pierre, P. (2014). Protein synthesis regulation, a pillar of strength for innate immunity? Curr. Opin. Immunol. 32, 28-35. Abstract Article
Artyukhin, A.B., Schroeder, F.C. and Avery, L. (2013). Density dependence in Caenorhabditis larval starvation. Sci. Rep. 3, 2777. Abstract Article
Ashe, A., Belicard, T., Le Pen, J., Sarkies, P., Frezal, L., Lehrbach, N.J., Félix, M.A., and Miska, E.A. (2013). A deletion polymorphism in the Caenorhabditis elegans RIG-I homolog disables viral RNA dicing and antiviral immunity. eLife 2, e00994. Abstract Article
Ausubel, F.M. (2005). Are innate immune signaling pathways in plants and animals conserved? Nat. Immunol. 6, 973-979. Abstract Article
Banyai, L., and Patthy, L. (1998). Amoebapore homologs of Caenorhabditis elegans. Biochim. Biophys. Acta 1429, 259-264. Abstract Article
Barrière, A., and Félix, M.A. (2005). High local genetic diversity and low outcrossing rate in Caenorhabditis elegans natural populations. Curr. Biol. 15, 1176-1184. Abstract Article
Barron, G.L. (1977). The Nematode-Destroying fungi, Topics in Mycobiology No. 1 (Guelph, Ont. Canada: Lancester press). Abstract Article
Barsyte, D., Lovejoy, D.A., and Lithgow, G.J. (2001). Longevity and heavy metal resistance in daf-2 and age-1 long-lived mutants of Caenorhabditis elegans. FASEB J. 15, 627-634. Abstract Article
Bischof, L.J., Kao, C.Y., Los, F.C., Gonzalez, M.R., Shen, Z., Briggs, S.P., van der Goot, F.G., and Aroian, R.V. (2008). Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 4, e1000176. Abstract Article
Boehnisch, C., Wong, D., Habig, M., Isermann, K., Michiels, N.K., Roeder, T., May, R.C., and Schulenburg, H. (2011). Protist-type lysozymes of the nematode Caenorhabditis elegans contribute to resistance against pathogenic Bacillus thuringiensis. PLoS One 6, e24619. Abstract Article
Bolz, D.D., Tenor, J.L., and Aballay, A. (2010). A conserved PMK-1/p38 MAPK is required in Caenorhabditis elegans tissue-specific immune response to Yersinia pestis infection. J. Biol. Chem. 285, 10832-10840. Abstract Article
Brandt, J.P., and Ringstad, N. (2015). Toll-like receptor signaling promotes development and function of sensory neurons required for a C. elegans pathogen-avoidance behavior. Curr. Biol. 25, 2228-2237. Abstract Article
Burkewitz, K., Choe, K.P., Lee, E.C., Deonarine, A., and Strange, K. (2012). Characterization of the proteostasis roles of glycerol accumulation, protein degradation and protein synthesis during osmotic stress in C. elegans. PLoS One 7, e34153. Abstract Article
Capra, E.J., Skrovanek, S.M., and Kruglyak, L. (2008). Comparative developmental expression profiling of two C. elegans isolates. PLoS One 3, e4055. Abstract Article
Chavez, V., Mohri-Shiomi, A., and Garsin, D.A. (2009). Ce-Duox1/BLI-3 generates reactive oxygen species as a protective innate immune mechanism in Caenorhabditis elegans. Infect. Immun. 77, 4983-4989. Abstract Article
Choe, K.P., and Strange, K. (2008). Genome-wide RNAi screen and in vivo protein aggregation reporters identify degradation of damaged proteins as an essential hypertonic stress response. Am. J. Physiol. Cell Physiol. 295, C1488-1498. Abstract Article
Chou, T.C., Chiu, H.C., Kuo, C.J., Wu, C.M., Syu, W.J., Chiu, W.T., and Chen, C.S. (2013). Enterohaemorrhagic Escherichia coli O157:H7 Shiga-like toxin 1 is required for full pathogenicity and activation of the p38 mitogen-activated protein kinase pathway in Caenorhabditis elegans. Cell. Microbiol. 15, 82-97. Abstract Article
Christophides, G.K., Zdobnov, E., Barillas-Mury, C., Birney, E., Blandin, S., Blass, C., Brey, P.T., Collins, F.H., Danielli, A., Dimopoulos, G., et al. (2002). Immunity-related genes and gene families in Anopheles gambiae. Science 298, 159-165. Abstract Article
Chuang, C.F., and Bargmann, C.I. (2005). A Toll-interleukin 1 repeat protein at the synapse specifies asymmetric odorant receptor expression via ASK1 MAPKKK signaling. Genes Dev. 19, 270-281. Abstract Article
Cipollo, J.F., Awad, A.M., Costello, C.E., and Hirschberg, C.B. (2004). srf-3, a mutant of Caenorhabditis elegans, resistant to bacterial infection and to biofilm binding, is deficient in glycoconjugates. J. Biol. Chem. 279, 52893-52903. Abstract Article
Claudio, N., Dalet, A., Gatti, E., and Pierre, P. (2013). Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J. 32, 1214-1224. Abstract Article
Cohen, L.B., and Troemel, E.R. (2015). Microbial pathogenesis and host defense in the nematode C. elegans. Curr. Opin. Microbiol. 23, 94-101. Abstract Article
Coolon, J.D., Jones, K.L., Todd, T.C., Carr, B.C., and Herman, M.A. (2009). Caenorhabditis elegans genomic response to soil bacteria predicts environment-specific genetic effects on life history traits. PLoS Genet. 5, e1000503. Abstract Article
Couillault, C., Fourquet, P., Pophillat, M., and Ewbank, J.J. (2012). A UPR-independent infection-specific role for a BiP/GRP78 protein in the control of antimicrobial peptide expression in C. elegans epidermis. Virulence 3, 299-308. Abstract Article
Couillault, C., Pujol, N., Reboul, J., Sabatier, L., Guichou, J.F., Kohara, Y., and Ewbank, J.J. (2004). TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol. 5, 488-494. Abstract Article
Curt, A., Zhang, J., Minnerly, J., and Jia, K. (2014). Intestinal autophagy activity is essential for host defense against Salmonella typhimurium infection in Caenorhabditis elegans. Deve. Comp. Immunol. 45, 214-218. Abstract Article
Dai, L.L., Gao, J.X., Zou, C.G., Ma, Y.C., and Zhang, K.Q. (2015). mir-233 modulates the unfolded protein response in C. elegans during Pseudomonas aeruginosa infection. PLoS Pathog. 11, e1004606. Abstract Article
de Bono, M., and Bargmann, C.I. (1998). Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 94, 679-689. Abstract Article
Dierking, K., Polanowska, J., Omi, S., Engelmann, I., Gut, M., Lembo, F., Ewbank, J.J., and Pujol, N. (2011). Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe 9, 425-435. Abstract Article
Dijksterhuis, J., Veenhuis, M., and Harder, W. (1990). Ultrastructural study of adhesion and initial stages of infection of the nematode by conidia of Drechmeria coniospora. Mycol. Res. 94, 1-8. Abstract Article
Dishaw, L.J., Haire, R.N., and Litman, G.W. (2012). The amphioxus genome provides unique insight into the evolution of immunity. Brief. Funct. Genomics 11, 167-176. Abstract Article
Drace, K., McLaughlin, S., and Darby, C. (2009). Caenorhabditis elegans BAH-1 is a DUF23 protein expressed in seam cells and required for microbial biofilm binding to the cuticle. PLoS One 4, e6741. Abstract Article
Drechsler, C. (1941). Some hyphomycetes parasitic on free-living terricolous nematodes. Phytopathology 31, 773-802. Article
Drickamer, K., and Dodd, R.B. (1999). C-Type lectin-like domains in Caenorhabditis elegans: predictions from the complete genome sequence. Glycobiology 9, 1357-1369. Abstract Article
Du Pasquier, L. (2005). Meeting the demand for innate and adaptive immunities during evolution. Scand. J. Immunol. 62 Suppl 1, 39-48. Abstract Article
Dunbar, T.L., Yan, Z., Balla, K.M., Smelkinson, M.G., and Troemel, E.R. (2012). C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 11, 375-386. Abstract Article
Edens, W.A., Sharling, L., Cheng, G., Shapira, R., Kinkade, J.M., Lee, T., Edens, H.A., Tang, X., Sullards, C., Flaherty, D.B., et al. (2001). Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 154, 879-891. Abstract Article
Engelmann, I., Griffon, A., Tichit, L., Montanana-Sanchis, F., Wang, G., Reinke, V., Waterston, R.H., Hillier, L.W., and Ewbank, J.J. (2011). A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS One 6, e19055. Abstract Article
Engelmann, I., and Pujol, N. (2010). Innate immunity in C. elegans. Adv. Exp. Med. Biol. 708, 105-121. Abstract Article
Ermolaeva, M.A., Segref, A., Dakhovnik, A., Ou, H.L., Schneider, J.I., Utermöhlen, O., Hoppe, T., and Schumacher, B. (2013). DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature 501, 416-420. Abstract Article
Estes, K.A., Dunbar, T.L., Powell, J.R., Ausubel, F.M., and Troemel, E.R. (2010). bZIP transcription factor zip-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 107, 2153-2158. Abstract Article
Estes, K.A., Szumowski, S.C., and Troemel, E.R. (2011). Non-lytic, actin-based exit of intracellular parasites from C. elegans intestinal cells. PLoS Pathog. 7, e1002227. Abstract Article
Evans, E.A., Kawli, T., and Tan, M.W. (2008). Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog. 4, e1000175. Abstract Article
Ewbank, J.J. (2002). Tackling both sides of the host-pathogen equation with Caenorhabditis elegans. Microbes Infect. 4, 247-256. Abstract Article
Ewbank, J.J. Signaling in the immune response (January 23, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.83.1, http://www.wormbook.org. Abstract Article
Ewbank, J.J., and Pujol, N. (2010). Cellular homeostasis: coping with ER overload during an immune response. Curr. Biol. 20, R452-455. Abstract Article
Félix, M.A., Ashe, A., Piffaretti, J., Wu, G., Nuez, I., Bélicard, T., Jiang, Y., Zhao, G., Franz, C.J., Goldstein, L.D., et al. (2011). Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol. 9, e1000586. Abstract Article
Félix, M.A., and Braendle, C. (2010). The natural history of Caenorhabditis elegans. Curr. Biol. 20, R965-969. Abstract Article
Félix, M.A., and Duveau, F. (2012). Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae. BMC Biol. 10, 59. Abstract Article
Fukushige, T., Hawkins, M.G., and McGhee, J.D. (1998). The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev. Biol. 198, 286-302. Abstract Article
Garsin, D.A., Villanueva, J.M., Begun, J., Kim, D.H., Sifri, C.D., Calderwood, S.B., Ruvkun, G., and Ausubel, F.M. (2003). Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300, 1921. Abstract Article
George-Raizen, J.B., Shockley, K.R., Trojanowski, N.F., Lamb, A.L., and Raizen, D.M. (2014). Dynamically-expressed prion-like proteins form a cuticle in the pharynx of Caenorhabditis elegans. Biol. Open 3, 1139-1149. Abstract Article
Gravato-Nobre, M.J., Nicholas, H.R., Nijland, R., O'Rourke, D., Whittington, D.E., Yook, K.J., and Hodgkin, J. (2005). Multiple genes affect sensitivity of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum. Genetics 171, 1033-1045. Abstract Article
Gravato-Nobre, M.J., Stroud, D., O'Rourke, D., Darby, C., and Hodgkin, J. (2011). Glycosylation genes expressed in seam cells determine complex surface properties and bacterial adhesion to the cuticle of Caenorhabditis elegans. Genetics 187, 141-155. Abstract Article
Gürtler, C., Carty, M., Kearney, J., Schattgen, S.A., Ding, A., Fitzgerald, K.A., and Bowie, A.G. (2014). SARM regulates CCL5 production in macrophages by promoting the recruitment of transcription factors and RNA polymerase II to the Ccl5 promoter. J. Immunol. 192, 4821-4832. Abstract Article
Ha, E.M., Oh, C.T., Bae, Y.S., and Lee, W.J. (2005). A direct role for dual oxidase in Drosophila gut immunity. Science 310, 847-850. Abstract Article
Hammell, C.M., Karp, X., and Ambros, V. (2009). A feedback circuit involving let-7-family miRNAs and DAF-12 integrates environmental signals and developmental timing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 106, 18668-18673. Abstract Article
Haskins, K.A., Russell, J.F., Gaddis, N., Dressman, H.K., and Aballay, A. (2008). Unfolded protein response genes regulated by CED-1 are required for Caenorhabditis elegans innate immunity. Dev. Cell 15, 87-97. Abstract Article
Hattori, A., Mizuno, T., Akamatsu, M., Hisamoto, N., and Matsumoto, K. (2013). The Caenorhabditis elegans JNK signaling pathway activates expression of stress response genes by derepressing the Fos/HDAC repressor complex. PLoS Genet. 9, e1003315. Abstract Article
Henderson, S.T., and Johnson, T.E. (2001). daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 11, 1975-1980. Abstract Article
Hertweck, M., Gobel, C., and Baumeister, R. (2004). C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev. Cell 6, 577-588. Abstract Article
Hodgkin, J., Félix, M.A., Clark, L.C., Stroud, D., and Gravato-Nobre, M.J. (2013). Two Leucobacter strains exert complementary virulence on Caenorhabditis including death by worm-star formation. Curr. Biol. 23, 2157-2161. Abstract Article
Hodgkin, J., Kuwabara, P.E., and Corneliussen, B. (2000). A novel bacterial pathogen, Microbacterium nematophilum, induces morphological change in the nematode C. elegans. Curr. Biol. 10, 1615-1618. Abstract Article
Hoeckendorf, A., Stanisak, M., and Leippe, M. (2012). The saposin-like protein SPP-12 is an antimicrobial polypeptide in the pharyngeal neurons of Caenorhabditis elegans and participates in defence against a natural bacterial pathogen. Biochem. J. 445, 205-212. Abstract Article
Hoeven, R., McCallum, K.C., Cruz, M.R., and Garsin, D.A. (2011). Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog. 7, e1002453. Abstract Article
Höflich, J., Berninsone, P., Göbel, C., Gravato-Nobre, M.J., Libby, B.J., Darby, C., Politz, S.M., Hodgkin, J., Hirschberg, C.B., and Baumeister, R. (2004). Loss of srf-3-encoded nucleotide sugar transporter activity in Caenorhabditis elegans alters surface antigenicity and prevents bacterial adherence. J. Biol. Chem. 279, 30440-30448. Abstract Article
Hsu, A.L., Murphy, C.T., and Kenyon, C. (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142-1145. Abstract Article
Huang, G., Shi, L.Z., and Chi, H. (2009). Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine 48, 161-169. Abstract Article
Huffman, D.L., Abrami, L., Sasik, R., Corbeil, J., van der Goot, F.G., and Aroian, R.V. (2004). Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. U. S. A. 101, 10995-11000. Abstract Article
Inoue, H., Hisamoto, N., An, J.H., Oliveira, R.P., Nishida, E., Blackwell, T.K., and Matsumoto, K. (2005). The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 19, 2278-2283. Abstract Article
Iordanov, M.S., Pribnow, D., Magun, J.L., Dinh, T.H., Pearson, J.A., Chen, S.L., and Magun, B.E. (1997). Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 17, 3373-3381. Abstract Article
Irazoqui, J.E., Troemel, E.R., Feinbaum, R.L., Luhachack, L.G., Cezairliyan, B.O., and Ausubel, F.M. (2010a). Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 6, e1000982. Abstract Article
Irazoqui, J.E., Urbach, J.M., and Ausubel, F.M. (2010b). Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat. Rev. Immunol. 10, 47-58. Abstract Article
Janeway, C.A., Jr. (1998). Presidential Address to The American Association of Immunologists. The road less traveled by: the role of innate immunity in the adaptive immune response. J. Immunol. 161, 539-544. Abstract Article
Janeway, C.A., Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197-216. Abstract Article
Kaplan, F., Badri, D.V., Zachariah, C., Ajredini, R., Sandoval, F.J., Roje, S., Levine, L.H., Zhang, F., Robinette, S.L., Alborn, H.T., et al. (2009). Bacterial attraction and quorum sensing inhibition in Caenorhabditis elegans exudates. J. Chem. Ecol. 35, 878-892. Abstract Article
Kao, C.Y., Los, F.C., Huffman, D.L., Wachi, S., Kloft, N., Husmann, M., Karabrahimi, V., Schwartz, J.L., Bellier, A., Ha, C., et al. (2011). Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 7, e1001314. Abstract Article
Kato, Y., Aizawa, T., Hoshino, H., Kawano, K., Nitta, K., and Zhang, H. (2002). abf-1 and abf-2, ASABF-type antimicrobial peptide genes in Caenorhabditis elegans. Biochem. J. 361, 221-230. Abstract Article
Kawli, T., He, F., and Tan, M.W. (2010a). It takes nerves to fight infections: insights on neuro-immune interactions from C. elegans. Dis. Model. Mech. 3, 721-731. Abstract Article
Kawli, T., and Tan, M.W. (2008). Neuroendocrine signals modulate the innate immunity of Caenorhabditis elegans through insulin signaling. Nat. Immunol. 9, 1415-1424. Abstract Article
Kawli, T., Wu, C., and Tan, M.W. (2010b). Systemic and cell intrinsic roles of Gqα signaling in the regulation of innate immunity, oxidative stress, and longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 107, 13788-13793. Abstract Article
Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461-464. Abstract Article
Keshet, Y., and Seger, R. (2010). The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 661, 3-38. Abstract Article
Kim, D.H. (2013). Bacteria and the aging and longevity of Caenorhabditis elegans. Annu. Rev. Genet. 47, 233-246. Abstract Article
Kim, D.H., Feinbaum, R., Alloing, G., Emerson, F.E., Garsin, D.A., Inoue, H., Tanaka-Hino, M., Hisamoto, N., Matsumoto, K., Tan, M.W., et al. (2002). A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623-626. Abstract Article
Kim, D.H., Liberati, N.T., Mizuno, T., Inoue, H., Hisamoto, N., Matsumoto, K., and Ausubel, F.M. (2004). Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc. Natl. Acad. Sci. U. S. A. 101, 10990-10994. Abstract Article
Kimura, K.D., Tissenbaum, H.A., Liu, Y., and Ruvkun, G. (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942-946. Abstract Article
Kirienko, N.V., Ausubel, F.M., and Ruvkun, G. (2015). Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 112, 1821-1826. Abstract Article
Kondo, M., Yanase, S., Ishii, T., Hartman, P.S., Matsumoto, K., and Ishii, N. (2005). The p38 signal transduction pathway participates in the oxidative stress-mediated translocation of DAF-16 to Caenorhabditis elegans nuclei. Mech. Ageing Dev. 126, 642-647. Abstract Article
Kudlow, B.A., Zhang, L., and Han, M. (2012). Systematic analysis of tissue-restricted miRISCs reveals a broad role for microRNAs in suppressing basal activity of the C. elegans pathogen response. Mol. Cell 46, 530-541. Abstract Article
Kurz, C.L., and Ewbank, J.J. (2003). Caenorhabditis elegans: an emerging genetic model for the study of innate immunity. Nat. Rev. Genet. 4, 380-390. Abstract Article
Kurz, C.L., and Ewbank, J.J. (2007). Infection in a dish: high-throughput analyses of bacterial pathogenesis. Curr. Opin. Microbiol. 10, 10-16. Abstract Article
Labed, S.A., Omi, S., Gut, M., Ewbank, J.J., and Pujol, N. (2012). The pseudokinase NIPI-4 is a novel regulator of antimicrobial peptide gene expression. PLoS One 7, e33887. Abstract Article
Labrousse, A., Chauvet, S., Couillault, C., Kurz, C.L., and Ewbank, J.J. (2000). Caenorhabditis elegans is a model host for Salmonella typhimurium. Curr. Biol. 10, 1543-1545. Abstract Article
Laing, S.T., Ivens, A., Butler, V., Ravikumar, S.P., Laing, R., Woods, D.J., and Gilleard, J.S. (2012). The transcriptional response of Caenorhabditis elegans to ivermectin exposure identifies novel genes involved in the response to reduced food intake. PLoS One 7, e31367. Abstract Article
Lamitina, S.T., and Strange, K. (2004). Transcriptional targets of the DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am. J. Physiol. Cell Physiol. 288, C467-474. Abstract Article
Landis, J.N., and Murphy, C.T. (2010). Integration of diverse inputs in the regulation of Caenorhabditis elegans DAF-16/FOXO. Dev. Dyn. 239, 1405-1412. Abstract Article
Lee, E.C., and Strange, K. (2012). GCN-2 dependent inhibition of protein synthesis activates osmosensitive gene transcription via WNK and Ste20 kinase signaling. Am. J. Physiol. Cell Physiol. 303, C1269-1277. Abstract Article
Lee, K., Shim, J., Bae, J., Kim, Y.J., and Lee, J. (2012). Stabilization of RNT-1 protein, runt-related transcription factor (RUNX) protein homolog of Caenorhabditis elegans, by oxidative stress through mitogen-activated protein kinase pathway. J. Biol. Chem. 287, 10444-10452. Abstract Article
Lee, K.A., Kim, S.H., Kim, E.K., Ha, E.M., You, H., Kim, B., Kim, M.J., Kwon, Y., Ryu, J.H., and Lee, W.J. (2013a). Bacterial-derived uracil as a modulator of mucosal immunity and gut-microbe homeostasis in Drosophila. Cell 153, 797-811. Abstract Article
Lee, S.H., Wong, R.R., Chin, C.Y., Lim, T.Y., Eng, S.A., Kong, C., Ijap, N.A., Lau, M.S., Lim, M.P., Gan, Y.H., et al. (2013b). Burkholderia pseudomallei suppresses Caenorhabditis elegans immunity by specific degradation of a GATA transcription factor. Proc. Natl. Acad. Sci. U. S. A. 110, 15067-15072. Abstract Article
Leroy, M., Mosser, T., Maniere, X., Alvarez, D.F., and Matic, I. (2012). Pathogen-induced Caenorhabditis elegans developmental plasticity has a hormetic effect on the resistance to biotic and abiotic stresses. BMC Evol. Biol. 12, 187. Abstract Article
Liang, B., Moussaif, M., Kuan, C.J., Gargus, J.J., and Sze, J.Y. (2006). Serotonin targets the DAF-16/FOXO signaling pathway to modulate stress responses. Cell Metab. 4, 429-440. Abstract Article
Liberati, N.T., Fitzgerald, K.A., Kim, D.H., Feinbaum, R., Golenbock, D.T., and Ausubel, F.M. (2004). Requirement for a conserved Toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc. Natl. Acad. Sci. U. S. A. 101, 6593-6598. Abstract Article
Liu, F., He, C.X., Luo, L.J., Zou, Q.L., Zhao, Y.X., Saini, R., Han, S.F., Knolker, H.J., Wang, L.S., and Ge, B.X. (2013). Nuclear hormone receptor regulation of microRNAs controls innate immune responses in C. elegans. PLoS Pathog. 9, e1003545. Abstract Article
Liu, Y., Samuel, B.S., Breen, P.C., and Ruvkun, G. (2014). Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508, 406-410. Abstract Article
Ludewig, A.H., Izrayelit, Y., Park, D., Malik, R.U., Zimmermann, A., Mahanti, P., Fox, B.W., Bethke, A., Doering, F., Riddle, D.L., et al. (2013). Pheromone sensing regulates Caenorhabditis elegans lifespan and stress resistance via the deacetylase SIR-2.1. Proc. Natl. Acad. Sci. U. S. A. 110, 5522-5527. Abstract Article
Ludewig AH. and Schroeder FC. Ascaroside signaling in C. elegans (January 18, 2013), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.155.1, http://www.wormbook.org. Abstract Article
Maguire, S.M., Clark, C.M., Nunnari, J., Pirri, J.K., and Alkema, M.J. (2011). The C. elegans touch response facilitates escape from predacious fungi. Curr. Biol. 21, 1326-1330. Abstract Article
Mahajan-Miklos, S., Tan, M.W., Rahme, L.G., and Ausubel, F.M. (1999). Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96, 47-56. Abstract Article
Mallo, G.V., Kurz, C.L., Couillault, C., Pujol, N., Granjeaud, S., Kohara, Y., and Ewbank, J.J. (2002). Inducible antibacterial defense system in C. elegans. Curr. Biol. 12, 1209-1214. Abstract Article
Mansisidor, A.R., Cecere, G., Hoersch, S., Jensen, M.B., Kawli, T., Kennedy, L.M., Chavez, V., Tan, M.W., Lieb, J.D., and Grishok, A. (2011). A conserved PHD finger protein and endogenous RNAi modulate insulin signaling in Caenorhabditis elegans. PLoS Genet. 7, e1002299. Abstract Article
Marsh, E.K., van den Berg, M.C., and May, R.C. (2011). A two-gene balance regulates Salmonella typhimurium tolerance in the nematode Caenorhabditis elegans. PLoS One 6, e16839. Abstract Article
Matsuzawa, A., Saegusa, K., Noguchi, T., Sadamitsu, C., Nishitoh, H., Nagai, S., Koyasu, S., Matsumoto, K., Takeda, K., and Ichijo, H. (2005). ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 6, 587-592. Abstract Article
Matzinger, P. (2002). The danger model: a renewed sense of self. Science 296, 301-305. Abstract Article
McElwee, J., Bubb, K., and Thomas, J.H. (2003). Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell 2, 111-121. Abstract Article
McElwee, J.J., Schuster, E., Blanc, E., Thomas, J.H., and Gems, D. (2004). Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 279, 44533-44543. Abstract Article
McEwan, D.L., Kirienko, N.V., and Ausubel, F.M. (2012). Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 11, 364-374. Abstract Article
McGhee, J.D., Fukushige, T., Krause, M.W., Minnema, S.E., Goszczynski, B., Gaudet, J., Kohara, Y., Bossinger, O., Zhao, Y., Khattra, J., et al. (2009). ELT-2 is the predominant transcription factor controlling differentiation and function of the C. elegans intestine, from embryo to adult. Dev. Biol. 327, 551-565. Abstract Article
McGrath, P.T., Rockman, M.V., Zimmer, M., Jang, H., Macosko, E.Z., Kruglyak, L., and Bargmann, C.I. (2009). Quantitative mapping of a digenic behavioral trait implicates globin variation in C. elegans sensory behaviors. Neuron 61, 692-699. Abstract Article
McMullan, R., Anderson, A., and Nurrish, S. (2012). Behavioral and immune responses to infection require Gαq- RhoA signaling in C. elegans. PLoS Pathog. 8, e1002530. Abstract Article
Means, T.K., Mylonakis, E., Tampakakis, E., Colvin, R.A., Seung, E., Puckett, L., Tai, M.F., Stewart, C.R., Pukkila-Worley, R., Hickman, S.E., et al. (2009). Evolutionarily conserved recognition and innate immunity to fungal pathogens by the scavenger receptors SCARF1 and CD36. J. Exp. Med. 206, 637-653. Abstract Article
Medzhitov, R., and Janeway, C.A., Jr. (1998). An ancient system of host defense. Curr. Opin. Immunol. 10, 12-15. Abstract Article
Medzhitov, R., Schneider, D.S., and Soares, M.P. (2012). Disease tolerance as a defense strategy. Science 335, 936-941. Abstract Article
Melo, J.A., and Ruvkun, G. (2012). Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149, 452-466. Abstract Article
Miller, E.V., Grandi, L.N., Giannini, J.A., Robinson, J.D., and Powell, J.R. (2015). The conserved G-protein coupled receptor FSHR-1 regulates protective host responses to infection and oxidative otress. PLoS One 10, e0137403. Abstract Article
Miltsch, S.M., Seeberger, P.H., and Lepenies, B. (2014). The C-type lectin-like domain containing proteins Clec-39 and Clec-49 are crucial for Caenorhabditis elegans immunity against Serratia marcescens infection. Dev. Comp. Immunol. 45, 67-73. Abstract Article
Mizuno, T., Hisamoto, N., Terada, T., Kondo, T., Adachi, M., Nishida, E., Kim, D.H., Ausubel, F.M., and Matsumoto, K. (2004). The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 23, 2226-2234. Abstract Article
Mochii, M., Yoshida, S., Morita, K., Kohara, Y., and Ueno, N. (1999). Identification of transforming growth factor-β-regulated genes in Caenorhabditis elegans by differential hybridization of arrayed cDNAs. Proc. Natl. Acad. Sci. U. S. A. 96, 15020-15025. Abstract Article
Mohr, I., and Sonenberg, N. (2012). Host translation at the nexus of infection and immunity. Cell Host Microbe 12, 470-483. Abstract Article
Moribe, H., Konakawa, R., Koga, D., Ushiki, T., Nakamura, K., and Mekada, E. (2012). Tetraspanin is required for generation of reactive oxygen species by the dual oxidase system in Caenorhabditis elegans. PLoS Genet. 8, e1002957. Abstract Article
Muhammed, M., Fuchs, B.B., Wu, M.P., Breger, J., Coleman, J.J., and Mylonakis, E. (2012). The role of mycelium production and a MAPK-mediated immune response in the C. elegans-Fusarium model system. Med. Mycol. 50, 488-496. Abstract Article
Murphy, C.T., McCarroll, S.A., Bargmann, C.I., Fraser, A., Kamath, R.S., Ahringer, J., Li, H., and Kenyon, C. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277-283. Abstract Article
Nathoo, A.N., Moeller, R.A., Westlund, B.A., and Hart, A.C. (2001). Identification of neuropeptide-like protein gene families in Caenorhabditis elegans and other species. Proc. Natl. Acad. Sci. U. S. A. 98, 14000-14005. Abstract Article
Nicholas, H.R., and Hodgkin, J. (2004a). The ERK MAP kinase cascade mediates tail swelling and a protective response to rectal infection in C. elegans. Curr. Biol. 14, 1256-1261. Abstract Article
Nicholas, H.R., and Hodgkin, J. (2004b). Responses to infection and possible recognition strategies in the innate immune system of Caenorhabditis elegans. Mol. Immunol. 41, 479-493. Abstract Article
O'Rourke, D., Baban, D., Demidova, M., Mott, R., and Hodgkin, J. (2006). Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum. Genome Res. 16, 1005-1016. Abstract Article
Oh, S.W., Mukhopadhyay, A., Dixit, B.L., Raha, T., Green, M.R., and Tissenbaum, H.A. (2006). Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat. Genet. 38, 251-257. Abstract Article
Olofsson, B. (2014). The olfactory neuron AWC promotes avoidance of normally palatable food following chronic dietary restriction. J. Exp. Biol. 217, 1790-1798. Abstract Article
Ookuma, S., Fukuda, M., and Nishida, E. (2003). Identification of a DAF-16 transcriptional target gene, scl-1, that regulates longevity and stress resistance in Caenorhabditis elegans. Curr. Biol. 13, 427-431. Abstract Article
Osterloh, J.M., Yang, J., Rooney, T.M., Fox, A.N., Adalbert, R., Powell, E.H., Sheehan, A.E., Avery, M.A., Hackett, R., Logan, M.A., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481-484. Abstract Article
Palaima, E., Leymarie, N., Stroud, D., Mizanur, R.M., Hodgkin, J., Gravato-Nobre, M.J., Costello, C.E., and Cipollo, J.F. (2010). The Caenorhabditis elegans bus-2 mutant reveals a new class of O-glycans affecting bacterial resistance. J. Biol. Chem. 285, 17662-17672. Abstract Article
Papp, D., Csermely, P., and Soti, C. (2012). A role for SKN-1/Nrf in pathogen resistance and immunosenescence in Caenorhabditis elegans. PLoS Pathog. 8, e1002673. Abstract Article
Parsons, L.M., and Cipollo, J. (2014). Oral ingestion of Microbacterium nematophilum leads to anal-region infection in Caenorhabditis elegans. Microbes Infect. 16, 356-361. Abstract Article
Parsons, L.M., Mizanur, R.M., Jankowska, E., Hodgkin, J., D, O.R., Stroud, D., Ghosh, S., and Cipollo, J.F. (2014). Caenorhabditis elegans bacterial pathogen resistant bus-4 mutants produce altered mucins. PLoS One 9, e107250. Abstract Article
Partridge, F.A., Tearle, A.W., Gravato-Nobre, M.J., Schafer, W.R., and Hodgkin, J. (2008). The C. elegans glycosyltransferase BUS-8 has two distinct and essential roles in epidermal morphogenesis. Dev. Biol. 317, 549-559. Abstract Article
Pellegrino, M.W., Nargund, A.M., Kirienko, N.V., Gillis, R., Fiorese, C.J., and Haynes, C.M. (2014). Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516, 414-417. Abstract Article
Peng, J., Yuan, Q., Lin, B., Panneerselvam, P., Wang, X., Luan, X.L., Lim, S.K., Leung, B.P., Ho, B., and Ding, J.L. (2010). SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur. J. Immunol. 40, 1738-1747. Abstract Article
Posas, F., and Saito, H. (1997). Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 276, 1702-1705. Abstract Article
Powell, J.R., and Ausubel, F.M. (2008). Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol. Biol., 415, 403-427. Abstract Article
Powell, J.R., Kim, D.H., and Ausubel, F.M. (2009). The G protein-coupled receptor FSHR-1 is required for the Caenorhabditis elegans innate immune response. Proc. Natl. Acad. Sci. U. S. A. 106, 2782-2787. Abstract Article
Pradel, E., Zhang, Y., Pujol, N., Matsuyama, T., Bargmann, C.I., and Ewbank, J.J. (2007). Detection and avoidance of a natural product from the pathogenic bacterium Serratia marcescens by Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 104, 2295-2300. Abstract Article
Pujol, N., Cypowyj, S., Ziegler, K., Millet, A., Astrain, A., Goncharov, A., Jin, Y., Chisholm, A.D., and Ewbank, J.J. (2008a). Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr. Biol. 18, 481-489. Abstract Article
Pujol, N., Davis, P.A., and Ewbank, J.J. (2012). The origin and function of anti-fungal peptides in C. elegans: open questions. Front. Immunol. 3, 237. Abstract Article
Pujol, N., Link, E.M., Liu, L.X., Kurz, C.L., Alloing, G., Tan, M.W., Ray, K.P., Solari, R., Johnson, C.D., and Ewbank, J.J. (2001). A reverse genetic analysis of components of the Toll signalling pathway in Caenorhabditis elegans. Curr. Biol. 11, 809-821. Abstract Article
Pujol, N., Zugasti, O., Wong, D., Couillault, C., Kurz, C.L., Schulenburg, H., and Ewbank, J.J. (2008b). Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 4, e1000105. Abstract Article
Pukkila-Worley, R., Ausubel, F.M., and Mylonakis, E. (2011). Candida albicans infection of Caenorhabditis elegans induces antifungal immune defenses. PLoS Pathog. 7, e1002074. Abstract Article
Pukkila-Worley, R., Feinbaum, R.L., McEwan, D.L., Conery, A.L., and Ausubel, F.M. (2014). The evolutionarily conserved mediator subunit MDT-15/MED15 links protective innate immune responses and xenobiotic detoxification. PLoS Pathog. 10, e1004143. Abstract Article
Reddy, K.C., Andersen, E.C., Kruglyak, L., and Kim, D.H. (2009). A polymorphism in npr-1 is a behavioral determinant of pathogen susceptibility in C. elegans. Science 323, 382-384. Abstract Article
Ren, M., Feng, H., Fu, Y., Land, M., and Rubin, C.S. (2009). Protein kinase D is an essential regulator of C. elegans innate immunity. Immunity 30, 521-532. Abstract Article
Ren, Z., and Ambros, V.R. (2015). Caenorhabditis elegans microRNAs of the let-7 family act in innate immune response circuits and confer robust developmental timing against pathogen stress. Proc. Natl. Acad. Sci. U. S. A. 112, E2366-2375. Abstract Article
Reynolds, R.M., and Phillips, P.C. (2013). Natural variation for lifespan and stress response in the nematode Caenorhabditis remanei. PLoS One 8, e58212. Abstract Article
Richardson, C.E., Kinkel, S., and Kim, D.H. (2011). Physiological IRE-1-XBP-1 and PEK-1 signaling in Caenorhabditis elegans larval development and immunity. PLoS Genet. 7, e1002391. Abstract Article
Richardson, C.E., Kooistra, T., and Kim, D.H. (2010). An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463, 1092-1095. Abstract Article
Roberts, A.F., Gumienny, T.L., Gleason, R.J., Wang, H., and Padgett, R.W. (2010). Regulation of genes affecting body size and innate immunity by the DBL-1/BMP-like pathway in Caenorhabditis elegans. BMC Dev. Biol. 10, 61. Abstract Article
Roeder, T., Stanisak, M., Gelhaus, C., Bruchhaus, I., Grotzinger, J., and Leippe, M. (2010). Caenopores are antimicrobial peptides in the nematode Caenorhabditis elegans instrumental in nutrition and immunity. Dev. Comp. Immunol. 34, 203-209. Abstract Article
Rohlfing, A.K., Miteva, Y., Hannenhalli, S., and Lamitina, T. (2010). Genetic and physiological activation of osmosensitive gene expression mimics transcriptional signatures of pathogen infection in C. elegans. PLoS One 5, e9010. Abstract Article
Rosso, M.N., Pujol, N., and Ewbank, J.J. (2013). Innate immunity in Caenorhabditis elegans and other nematodes. In Parasitic Nematodes: Molecular Biology, Biochemistry and Immunology, M.W. Kennedy, and W. Harnett, eds. (New York: CABI Publishing), pp. 42-66.
Rouger, V., Bordet, G., Couillault, C., Monneret, S., Mailfert, S., Ewbank, J.J., Pujol, N., and Marguet, D. (2014). Independent synchronized control and visualization of interactions between living cells and organisms. Biophys. J. 106, 2096-2104. Abstract Article
Runkel, E.D., Liu, S., Baumeister, R., and Schulze, E. (2013). Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 9, e1003346. Abstract Article
Sackton, T.B., Lazzaro, B.P., Schlenke, T.A., Evans, J.D., Hultmark, D., and Clark, A.G. (2007). Dynamic evolution of the innate immune system in Drosophila. Nat. Genet. 39, 1461-1468. Abstract Article
Sagasti, A., Hisamoto, N., Hyodo, J., Tanaka-Hino, M., Matsumoto, K., and Bargmann, C.I. (2001). The CaMKII UNC-43 activates the MAPKKK NSY-1 to execute a lateral signaling decision required for asymmetric olfactory neuron fates. Cell 105, 221-232. Abstract Article
Sahu, S.N., Lewis, J., Patel, I., Bozdag, S., Lee, J.H., LeClerc, J.E., and Cinar, H.N. (2012). Genomic analysis of immune response against Vibrio cholerae hemolysin in Caenorhabditis elegans. PLoS One 7, e38200. Abstract Article
Sarkies, P., Ashe, A., Le Pen, J., McKie, M.A., and Miska, E.A. (2013). Competition between virus-derived and endogenous small RNAs regulates gene expression in Caenorhabditis elegans. Genome Res. 23, 1258-1270. Abstract Article
Schulenburg, H., and Boehnisch, C. (2008). Diversification and adaptive sequence evolution of Caenorhabditis lysozymes (Nematoda: Rhabditidae). BMC Evol. Biol. 8, 114. Abstract Article
Schulenburg, H., and Ewbank, J.J. (2004). Diversity and specificity in the interaction between Caenorhabditis elegans and the pathogen Serratia marcescens. BMC Evol. Biol. 4, 49. Abstract Article
Schulenburg, H., and Ewbank, J.J. (2007). The genetics of pathogen avoidance in Caenorhabditis elegans. Mol. Microbiol. 66, 563-570. Abstract Article
Schulenburg, H., Hoeppner, M.P., Weiner, J., 3rd, and Bornberg-Bauer, E. (2008). Specificity of the innate immune system and diversity of C-type lectin domain (CTLD) proteins in the nematode Caenorhabditis elegans. Immunobiology 213, 237-250. Abstract Article
Sem, X., Kreisberg, J.F., Kawli, T., Tan, M.W., Rhen, M., and Tan, P. (2012). Modulation of Caenorhabditis elegans infection sensitivity by the LIN-7 cell junction protein. Cell. Microbiol. 14, 1584-1599. Abstract Article
Seo, K., Choi, E., Lee, D., Jeong, D.E., Jang, S.K., and Lee, S.J. (2013). Heat shock factor 1 mediates the longevity conferred by inhibition of TOR and insulin/IGF-1 signaling pathways in C. elegans. Aging Cell 12, 1073-1081. Abstract Article
Seong, S.Y., and Matzinger, P. (2004). Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 4, 469-478. Abstract Article
Shapira, M., Hamlin, B.J., Rong, J., Chen, K., Ronen, M., and Tan, M.W. (2006). A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103, 14086-14091. Abstract Article
Shivers, R.P., Pagano, D.J., Kooistra, T., Richardson, C.E., Reddy, K.C., Whitney, J.K., Kamanzi, O., Matsumoto, K., Hisamoto, N., and Kim, D.H. (2010). Phosphorylation of the conserved transcription factor ATF-7 by PMK-1 p38 MAPK regulates innate immunity in Caenorhabditis elegans. PLoS Genet. 6, e1000892. Abstract Article
Sifri, C.D., Begun, J., and Ausubel, F.M. (2005). The worm has turned—microbial virulence modeled in Caenorhabditis elegans. Trends Microbiol. 13, 119-127. Abstract Article
Sifri, C.D., Begun, J., Ausubel, F.M., and Calderwood, S.B. (2003). Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect. Immun. 71, 2208-2217. Abstract Article
Simonsen, K.T., Gallego, S.F., Faergeman, N.J., and Kallipolitis, B.H. (2012). Strength in numbers: “Omics” studies of C. elegans innate immunity. Virulence 3, 477-484. Abstract Article
Simonsen, K.T., Moller-Jensen, J., Kristensen, A.R., Andersen, J.S., Riddle, D.L., and Kallipolitis, B.H. (2011). Quantitative proteomics identifies ferritin in the innate immune response of C. elegans. Virulence 2, 120-130. Abstract Article
Singh, V., and Aballay, A. (2006). Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc. Natl. Acad. Sci. U. S. A. 103, 13092-13097. Abstract Article
Singh, V., and Aballay, A. (2009). Regulation of DAF-16-mediated innate immunity in Caenorhabditis elegans. J. Biol. Chem. 284, 35580-35587. Abstract Article
Sinha, A., Rae, R., Iatsenko, I., and Sommer, R.J. (2012). System wide analysis of the evolution of innate immunity in the nematode model species Caenorhabditis elegans and Pristionchus pacificus. PLoS One 7, e44255. Abstract Article
Solomon, A., Bandhakavi, S., Jabbar, S., Shah, R., Beitel, G.J., and Morimoto, R.I. (2004). Caenorhabditis elegans OSR-1 regulates behavioral and physiological responses to hyperosmotic environments. Genetics 167, 161-170. Abstract Article
Sommer, R.J., and McGaughran, A. (2013). The nematode Pristionchus pacificus as a model system for integrative studies in evolutionary biology. Mol. Ecol. 22, 2380-2393. Abstract Article
Styer, K.L., Singh, V., Macosko, E., Steele, S.E., Bargmann, C.I., and Aballay, A. (2008). Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science 322, 460-464. Abstract Article
Sullivan, J.C., Wolenski, F.S., Reitzel, A.M., French, C.E., Traylor-Knowles, N., Gilmore, T.D., and Finnerty, J.R. (2009). Two alleles of NF-κB in the sea anemone Nematostella vectensis are widely dispersed in nature and encode proteins with distinct activities. PLoS One 4, e7311. Abstract Article
Sun, J., Singh, V., Kajino-Sakamoto, R., and Aballay, A. (2011). Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332, 729-732. Abstract Article
Tan, M.W. (2002). Identification of host and pathogen factors involved in virulence using Caenorhabditis elegans. Methods Enzymol. 358, 13-28. Abstract Article
Tao, L., Xie, Q., Ding, Y.H., Li, S.T., Peng, S., Zhang, Y.P., Tan, D., Yuan, Z., and Dong, M.Q. (2013). CAMKII and calcineurin regulate the lifespan of Caenorhabditis elegans through the FOXO transcription factor DAF-16. eLife 2, e00518. Abstract Article
Thakur, N., Pujol, N., Tichit, L., and Ewbank, J.J. (2014). Clone mapper: an online suite of tools for RNAi experiments in Caenorhabditis elegans. G3 4, 2137-2145. Abstract Article
Thomas, J.H. (2006a). Adaptive evolution in two large families of ubiquitin-ligase adapters in nematodes and plants. Genome Res. 16, 1017-1030. Abstract Article
Thomas, J.H. (2006b). Concerted evolution of two novel protein families in Caenorhabditis species. Genetics 172, 2269-2281. Abstract Article
Tiller, G.R., and Garsin, D.A. (2014). The SKPO-1 peroxidase functions in the hypodermis to protect Caenorhabditis elegans from bacterial infection. Genetics 197, 515-526. Abstract Article
Tong, A., Lynn, G., Ngo, V., Wong, D., Moseley, S.L., Ewbank, J.J., Goncharov, A., Wu, Y.C., Pujol, N., and Chisholm, A.D. (2009). Negative regulation of Caenorhabditis elegans epidermal damage responses by death-associated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 106, 1457-1461. Abstract Article
Tonsaker, T., Pratt, R.M., and McGhee, J.D. (2012). Re-evaluating the role of ELT-3 in a GATA transcription factor circuit proposed to guide aging in C. elegans. Mech. Ageing Dev. 133, 50-53. Abstract Article
Treitz, C., Cassidy, L., Hockendorf, A., Leippe, M., and Tholey, A. (2015). Quantitative proteome analysis of Caenorhabditis elegans upon exposure to nematicidal Bacillus thuringiensis. J. Proteomics 113, 337-350. Abstract Article
Troemel, E.R., Chu, S.W., Reinke, V., Lee, S.S., Ausubel, F.M., and Kim, D.H. (2006). p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2, e183. Abstract Article
Troemel, E.R., Félix, M.A., Whiteman, N.K., Barrière, A., and Ausubel, F.M. (2008). Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol. 6, 2736-2752. Abstract Article
Troemel, E.R., Sagasti, A., and Bargmann, C.I. (1999). Lateral signaling mediated by axon contact and calcium entry regulates asymmetric odorant receptor expression in C. elegans. Cell 99, 387-398. Abstract Article
Tullet, J.M., Araiz, C., Sanders, M.J., Au, C., Benedetto, A., Papatheodorou, I., Clark, E., Schmeisser, K., Jones, D., Schuster, E.F., et al. (2014). DAF-16/FoxO directly regulates an atypical AMP-activated protein kinase gamma isoform to mediate the effects of insulin/IGF-1 signaling on aging in Caenorhabditis elegans. PLoS Genet. 10, e1004109. Abstract Article
Tullet, J.M., Hertweck, M., An, J.H., Baker, J., Hwang, J.Y., Liu, S., Oliveira, R.P., Baumeister, R., and Blackwell, T.K. (2008). Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132, 1025-1038. Abstract Article
Urano, F., Calfon, M., Yoneda, T., Yun, C., Kiraly, M., Clark, S.G., and Ron, D. (2002). A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J. Cell Biol. 158, 639-646. Abstract Article
Vaishnava, S., Yamamoto, M., Severson, K.M., Ruhn, K.A., Yu, X., Koren, O., Ley, R., Wakeland, E.K., and Hooper, L.V. (2011). The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255-258 Abstract Article
van den Berg, L.M., Gringhuis, S.I., and Geijtenbeek, T.B. (2012). An evolutionary perspective on C-type lectins in infection and immunity. Ann. N. Y. Acad. Sci. 1253, 149-158. Abstract Article
van der Hoeven, R., Cruz, M.R., Chavez, V., and Garsin, D.A. (2015). Localization of the dual oxidase BLI-3 and characterization of its NADPH oxidase domain during infection of Caenorhabditis elegans. PLoS One 10, e0124091. Abstract Article
Visvikis, O., Ihuegbu, N., Labed, S.A., Luhachack, L.G., Alves, A.M., Wollenberg, A.C., Stuart, L.M., Stormo, G.D., and Irazoqui, J.E. (2014). Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 40, 896-909. Abstract Article
Viswanathan, M., Kim, S.K., Berdichevsky, A., and Guarente, L. (2005). A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev. Cell 9, 605-615. Abstract Article
Volkers, R.J., Snoek, L.B., Hubar, C.J., Coopman, R., Chen, W., Yang, W., Sterken, M.G., Schulenburg, H., Braeckman, B.P., and Kammenga, J.E. (2013). Gene-environment and protein-degradation signatures characterize genomic and phenotypic diversity in wild Caenorhabditis elegans populations. BMC Biol. 11, 93. Abstract Article
von Reuss, S.H., Bose, N., Srinivasan, J., Yim, J.J., Judkins, J.C., Sternberg, P.W., and Schroeder, F.C. (2012). Comparative metabolomics reveals biogenesis of ascarosides, a modular library of small-molecule signals in C. elegans. J. Am. Chem. Soc. 134, 1817-1824. Abstract Article
Wang, J., Robida-Stubbs, S., Tullet, J.M., Rual, J.F., Vidal, M., and Blackwell, T.K. (2010). RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 6, e1001048. Abstract Article
Weiberg, A., Wang, M., Lin, F.M., Zhao, H., Zhang, Z., Kaloshian, I., Huang, H.D., and Jin, H. (2013). Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342, 118-123. Abstract Article
Wheeler, J.M., and Thomas, J.H. (2006). Identification of a novel gene family involved in osmotic stress response in Caenorhabditis elegans. Genetics 174, 1327-1336. Abstract Article
Witting, M., Lucio, M., Tziotis, D., Wagele, B., Suhre, K., Voulhoux, R., Garvis, S., and Schmitt-Kopplin, P. (2015). DI-ICR-FT-MS-based high-throughput deep metabotyping: a case study of the Caenorhabditis elegans-Pseudomonas aeruginosa infection model. Anal. Bioanal. Chem. 407, 1059-1073. Abstract Article
Wolff, S., Ma, H., Burch, D., Maciel, G.A., Hunter, T., and Dillin, A. (2006). SMK-1, an essential regulator of DAF-16-mediated longevity. Cell 124, 1039-1053. Abstract Article
Wong, D., Bazopoulou, D., Pujol, N., Tavernarakis, N., and Ewbank, J.J. (2007). Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol. 8, R194. Abstract Article
Xie, Y., Moussaif, M., Choi, S., Xu, L., and Sze, J.Y. (2013). RFX transcription factor DAF-19 regulates 5-HT and innate immune responses to pathogenic bacteria in Caenorhabditis elegans. PLoS Genet. 9, e1003324. Abstract Article
Xu, S., and Chisholm, A.D. (2011). A Gαq-Ca2+ signaling pathway promotes actin-mediated epidermal wound closure in C. elegans. Curr. Biol. 21, 1960-1967. Abstract Article
Yamada, K., Hirotsu, T., Matsuki, M., Butcher, R.A., Tomioka, M., Ishihara, T., Clardy, J., Kunitomo, H., and Iino, Y. (2010). Olfactory plasticity is regulated by pheromonal signaling in Caenorhabditis elegans. Science, 329, 1647-1650. Abstract Article
Yang, W., Dierking, K., Esser, D., Tholey, A., Leippe, M., Rosenstiel, P., and Schulenburg, H. (2015). Overlapping and unique signatures in the proteomic and transcriptomic responses of the nematode Caenorhabditis elegans toward pathogenic Bacillus thuringiensis. Dev. Comp. Immunol. 51, 1-9. Abstract Article
Yook, K., and Hodgkin, J. (2007). Mos1 mutagenesis reveals a diversity of mechanisms affecting response of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum. Genetics 175, 681-697. Abstract Article
Yu, H., Lai, H.-J., Lin, T.-W., Chen, C.-S., and Lo, S.J. (2015). Loss of DNase II function in the gonad is associated with a higher expression of antimicrobial genes in C. elegans. Biochem. J. 470, 145-154. Abstract Article
Zhang, L., Li, L., Guo, X., Litman, G.W., Dishaw, L.J., and Zhang, G. (2015a). Massive expansion and functional divergence of innate immune genes in a protostome. Sci. Rep. 5, 8693. Abstract Article
Zhang, P., Judy, M., Lee, S.J., and Kenyon, C. (2013). Direct and indirect gene regulation by a life-extending FOXO protein in C. elegans: roles for GATA factors and lipid gene regulators. Cell Metab. 17, 85-100. Abstract Article
Zhang, X., and Zhang, Y. (2012). DBL-1, a TGF-β, is essential for Caenorhabditis elegans aversive olfactory learning. Proc. Natl. Acad. Sci. U. S. A. 109, 17081-17086. Abstract Article
Zhang, Y., Li, W., Li, L., Li, Y., Fu, R., Zhu, Y., Li, J., Zhou, Y., Xiong, S., and Zhang, H. (2015b). Structural damage in the C. elegans epidermis causes release of STA-2 and induction of an innate immune response. Immunity 42, 309-320. Abstract Article
Zhang, Y., Lu, H., and Bargmann, C.I. (2005). Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature 438, 179-184. Abstract Article
Ziegler, K., Kurz, C.L., Cypowyj, S., Couillault, C., Pophillat, M., Pujol, N., and Ewbank, J.J. (2009). Antifungal innate immunity in C. elegans: PKCδ links G protein signaling and a conserved p38 MAPK cascade. Cell Host Microbe 5, 341-352. Abstract Article
*Edited by Iva Greenwald. Last revised September 16, 2015. Published in its final form August 14, 2018. This chapter should be cited as: Kim D.H. and Ewbank J.J. Signaling in the innate immune response. (August 14, 2018), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.83.2, http://www.wormbook.org.
Copyright: © 2016 Dennis H. Kim and Jonathan J. Ewbank. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: dhkim@mit.edu or ewbank@ciml.univ-mrs.fr
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.