Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergAs described in SNPs: introduction and two-point mapping single nucleotide polymorphisms (SNPs) provide an exceptionally powerful means for supplementing traditional mapping practices. SNP mapping is both conceptually and practically an extension of standard three-point mapping (see Three-point mapping with genetic markers), and will invariably require some previous genetic mapping of the mutation of interest. The specific strategy for mapping mutations by SNP analysis will depend somewhat on the nature of your mutant phenotype. In most instances, it is advantageous to generate a chromosome containing your mutation that is flanked by two easily discernable genetic markers such as dpy and unc. The genetic distance between the flanking markers should be as small as possible, something in the range of a few map units. However, some regions of the chromosome will require that you use markers that are separated by significantly greater distances. Although not optimal, this is still workable. The issue is that the farther apart the markers are, the more recombinants that will ultimately be needed to obtain the same level of mapping refinement. Still, we have successfully used markers as far apart as 12 map units to narrow a region down to a handful of genes. Ultimately, it is more important to use easily discernable markers than to shave off a few map units of distance. Another point is that it is useful to be able to pick recombinants from both directions if at all possible. At the very least, it doubles the number of useful recombinants on the plates and can provide an added level of security, provided all your data points are in agreement.
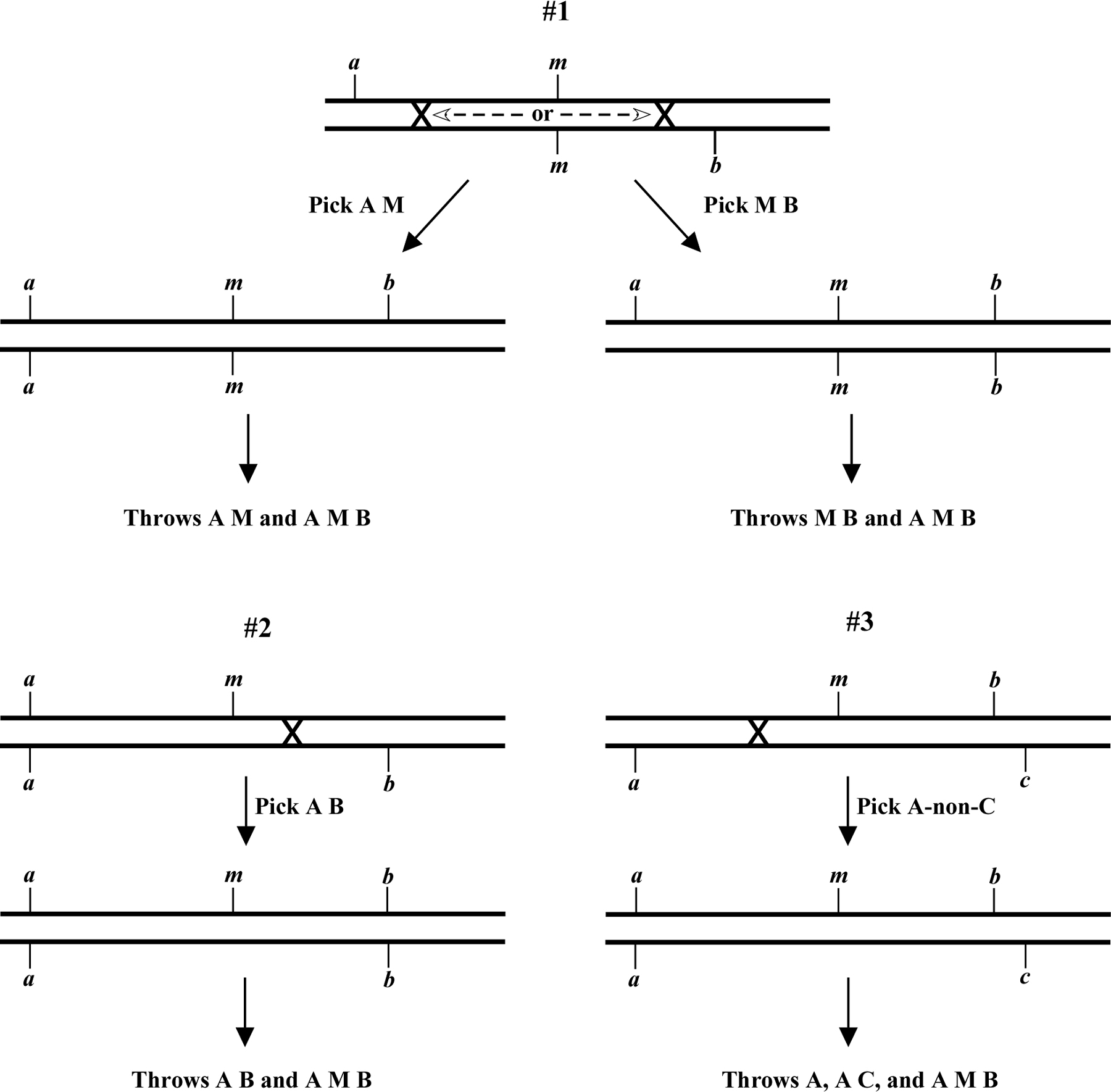
Creating strains for SNP mapping. For viable mutations, triple-mutant chromosomes can easily be obtained by generating balanced strains of the a m/m b genotype, where m is the mutation and a and b are the marker mutations (Figure 1, example #1). One then picks either A M or M B animals and looks for the appearance of A M B animals in some fraction of the self progeny (e.g., from a parent that was a m b/a m), indicating the presence of a recombinant a m b chromosome. The choice of picking A M or M B animals will depend on the ease of recognition of the A M B animal in either the A M or M B background. For example, it is somewhat easier to identify Dpy Unc animals from plates where most of other animals are Unc than the other way around.
For non-viable mutations, the construction and maintenance of the triple mutant chromosome is somewhat more involved. Here you will need to initially construct strains that are either a m/a b or m b/a b (Figure 1, example #2). Then by blindly picking sufficient animals of the A B phenotype, it should be possible to isolate strains that throw 1/4 A M B, indicating the a m b/a b genotype. Alternatively, you could generate the strain m b/a c, where c lies just to the right of b and confers a phenotype that is distinct from that of either a or b. By picking A-non-C animals, a reasonably high percentage of recombinants should be of the a m b/a c genotype (Figure 1, example #3). This approach has the added advantage that the strain generated is at least partially balanced and therefore easier to maintain.
On the other hand, if your mutation is inviable but you have already constructed several well-balanced strains that each carry the a m or the m b chromosomes, it is reasonable that you might want to use these strains directly for SNP mapping (also see below). The key here is to have two strains with markers on opposite sides, as this will allow you to move your region in from both directions. The same holds true for non-viable mutants when working with a triple-mutant chromosome (a m b). This is because the only recombinants that can be propagated as homozygotes (a/a or b/b) will necessarily have resulted from crossover events that lead to the loss of m. Thus, in this situation, it is essential to be able to pick both A-non-B and B-non-A animals to narrow down your region from both sides.
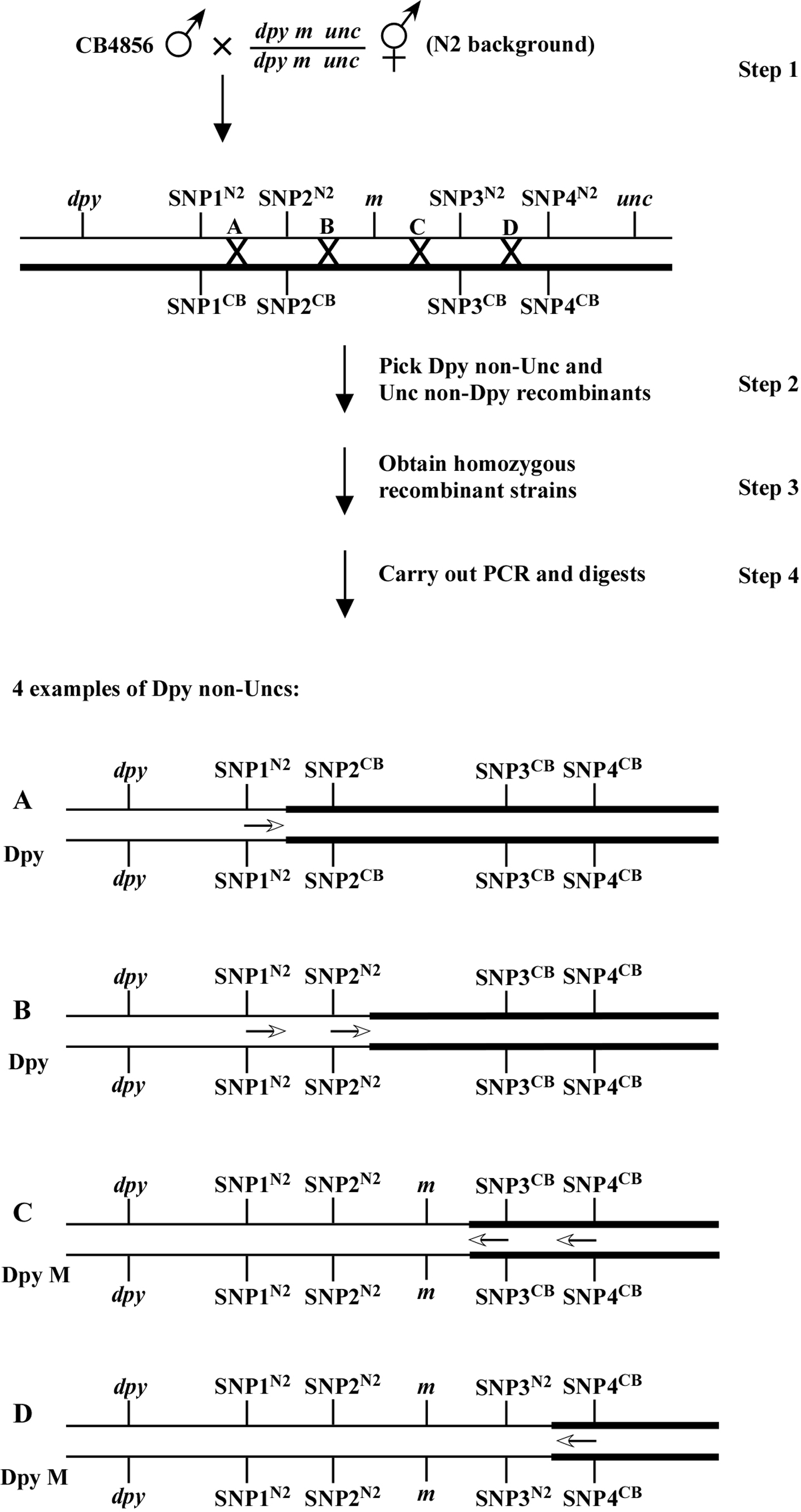
Figure 2 shows the basic flowchart and potential outcomes for mapping a viable mutation that is flanked by the visible markers a and b using four regional SNPs. In this scheme, F1 heterozygous hermaphrodites are generated through crosses and allowed to self (step 1). F2 recombinants are then isolated (step 2) and sufficient self progeny are subsequently cloned such that animals homozygous for the recombinant chromosome are obtained in the F3 generation (step 3). F4 progeny can then be assayed for the presence of N2 or CB4856 sequences at an given SNP locus (step 4). For example, a Dpy non-Unc resulting from a crossover at point A would be N2 for SNP1 and would not contain the mutation m. Thus, m must lie to right of SNP1. Likewise, a Dpy non-Unc resulting from a crossover at point C would result in an M animal that was CB4856 for SNP3 and SNP4. Thus, m must reside to the left of both SNP3 and SNP4. In the examples provided in Figure 2, the data from recombinants B and C establish the tightest endpoints on the left and right respectively. As described above, non-viable mutants may be most conveniently mapped using two strains, e.g., a m and m b (Figure 3). By picking A non-M from one strain and B non-M from the other, both sides can effectively be pushed in.
It is also worth noting that although we mostly use SNP mapping to generate progressively tighter endpoints, valuable three-point mapping data can be gleaned through this process. For example, imagine that you have assigned your mutation to the region between SNP2 and SNP3 (Figure 2). In doing so, you notice that of the 20 Dpy non-Unc recombinants that experienced a crossover in this region (meaning they are all N2 for SNP2 and CB4856 for SNP3), 18 contain the mutation m. This would suggest that m lies relatively close to SNP2, as the large majority of recombination events resulted in the retention of m on the dpy chromosome. Likewise, most Unc non-Dpy recombinants isolated in this region (meaning they are N2 for SNP3 and CB4856 for SNP2) would be expected to be non-M. This may at first seem counterintuitive. This is because for normal three-point mapping (Three-point mapping with genetic markers), the mutation is always on the opposite chromosome from the markers, hence a high percentage of Dpy-M animals would indicate that m lies closer to the unc side of things. The difference here is that the mutation and markers all start out on the same chromosome, thus a minor revision in thinking is required. As always, if this does not make immediate sense, draw it out.
The optimal strategies for efficient SNP mapping will vary somewhat depending on the nature of the mutant phenotype and on the time commitment of the researcher. We have had good success using a two-tiered approach. Initially, we isolate 75-100 recombinant animals and then in the subsequent generation isolate strains that are homozygous for the recombinant chromosome. We then begin the process of testing regional SNPs, initially being somewhat conservative in our choices of locations (meaning that we try not to overshoot). We then methodically move in our endpoints, discarding as we go those recombinants that are no longer informative. At some point, this initial pool will fail to yield any additional information and we are left with just a few recombinants holding down the left and right inner endpoints.
In the second phase of SNP mapping, we go for the "mother load", where several hundred or more recombinants are isolated. The strategy is to quickly screen these recombinants using the inner-most (or near inner-most) pair of informative SNPs. For reasons described above, it is preferable that these SNPs be CB4856-specific cutters, although well-behaved N2 cutters may be workable. Most important is that the chosen SNPs provide consistent and easily interpretable results. For carrying out PCR, you can use either the initial recombinant animal (after it has had 1-2 days to lay sufficient eggs) or 5 or 6 of its L4 progeny that display the recombinant phenotype.
To avert potential problems, it is advisable to stagger the growth of the recombinant animals as much as possible, either by picking recombinants over several days or by maintaining some at 20°C and others at 16°C. The idea is to avoid having just one or two "days from hell" when all the recombinants must be screened at once. Based on the position of the SNPs relative to the markers, we will test first one and then the other SNP for all of our recombinants. For example if one is testing Dpy non-Unc recombinants and the two innermost SNPs are located closer to the unc mutation, it will be most efficient to first test the proximal SNP (relative to dpy) to determine if it is N2, before bothering to test the distal SNP to see if it is CB4856. Non-informative recombinants are immediately pitched, whereas the progeny from the informative recombinants are further cloned to generate homozygous lines.
*Edited by Victor Ambros. Last revised May 4, 2005. Published February 17, 2006. This chapter should be cited as: Fay, D. Genetic mapping and manipulation: Chapter 5-SNPs: Three-point mapping (February 17, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.94.1, http://www.wormbook.org.
Copyright: © 2006 David Fay. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: davidfay@uwyo.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.