Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergMapping with SNPs has become a powerful complement (and in some cases outright alternative) to the standard genetic mapping procedures described above. In fact, the advent of SNP mapping has been nearly as significant for C. elegans forward genetics as RNAi has been for reverse genetics. With SNP mapping, basically all mutations are now theoretically clonable, something that wasn't true in the past. Moreover, SNP mapping can be routinely used to narrow down the known physical location of mutations to regions smaller than a single cosmid (∼30,000 bp; ∼6–7 genes). With genetic mapping, even in the best of circumstances, the implicated regions usually span 6–10 complete cosmids or more. In fact, SNP mapping can theoretically be used to narrow down the implicated region to a single gene, although this level of mapping is usually unnecessary and can become inordinately time consuming.
Although several approaches for mapping using polymorphisms have been described, we will focus here on the use the Hawaiian C. elegans isolate, CB4856 (Jakubowski and Kornfeld, 1999; Koch et al., 2000; Wicks et al., 2001; Swan et al., 2002). Geographical separation and several million years of evolutionary drift have led to a sizeable number of genetic differences (DNA polymorphisms) between the Hawaiian (CB4856) and English (N2) C. elegans isolates. In fact, differences in the genomic sequences of CB4856 and N2 occur on average every 1,000 base pairs. The majority of these changes occur in non-coding or intergenic regions and probably have no functional consequence. Some polymorphisms, however, clearly affect protein activity or gene expression, as N2 and CB4856 differ notably in a number of respects including their mating behaviors and relative sensitivities to RNAi (Tijsternman et al., 2002).
The term SNP is a bit of a misnomer. Although many of the sequence variations between N2 and CB4856 are indeed single-nucleotide changes (for example from an A to a G), small deletions or insertions are also very common. What is experimentally most relevant, however, is whether or not these polymorphisms affect the recognition site for an endonuclease. SNPs that result in restriction-fragment length polymorphisms (RFLPs; also called snip SNPs) are easier to work with, as digestion by enzymes is much more rapid and inexpensive than sending off samples for sequencing. Also, it generally doesn't matter whether it's the N2 or CB4856 DNA that is cleavable (however, also see Section 4), just so long as the digestion patterns of the two isolates are clearly distinguishable.
There are currently two C. elegans SNP databases accessible via the internet: http://genome.wustl.edu/genome/celegans/celegans_snp.cgi and http://genome.ucsc.edu/cgi-bin/hgTables?org=C.+elegans&db=ce4&hgsid=109019391&hgta_doMainPage=1.
The first was developed >5 years ago and contains only a partial listing of the many polymorphisms that exist between N2 and CB4856. Despite being incomplete, this database employs a straightforward interface and, for initial mapping purposes, is likely to be sufficient for most users. The newer database contains information that is somewhat more preliminary and the site is currently under development. However, since this database has been assembled using the complete sequence of CB4856, this site lists many more candidate SNPs than the original database. At present, the newer database may be best suited for finer mapping such as that commonly encountered in the later stages of 3-point SNP mapping or for determining endpoints using 2-point methods (described in Section 5). Both databases are described in some detail below.
The original C. elegans SNP database can be accessed at: http://genome.wustl.edu/genome/celegans/celegans_snp.cgi. Despite having only incomplete genome coverage, this resource provides a very useful inventory of many SNPs for the strains N2 and CB4856. This database is organized according to the physical map by chromosomes, chromosomal subsegments, and cosmids. For example, at the top of sequence Segment 9 on Chromosome X (click ‘Chromosome X Polymorphisms’ at the bottom of the page, then click ‘9’, or go to http://genome.wustl.edu/genome/celegans/chromX_layout.html then click on ‘9’), you will find the SNP B0403:33022 S=CT. This means the polymorphism is on cosmid B0403 at nucleotide position 33,022 and that the two strains differ in having either a C or T at this position. SNPs listed in red lettering have presumably been experimentally confirmed, whereas SNPs listed in white lettering are as yet unconfirmed. In fact, our lab has had at least one bad experience with a “confirmed” SNP, thus it is essential to make sure that any SNP you work with behaves as expected in your own hands.
Clicking on the red letters of B0403:33022 S=CT, we bring up an additional window that shows the actual sequences surrounding the SNP in black lettering (usually ∼500 bp upstream and downstream) as well as the SNP itself in red lettering [C/T]. This designation indicates that N2 contains a C at this position whereas CB4856 contains a T. Also, if it is an RFLP-type SNP, the top of this page will show predicted digestion sites for the displayed DNA sequence from N2 and CB4856 (listed here as “HA” for Hawaiian), using one or more enzymes. Looking at this, we notice that in the CB4856 background the presence of the T results in the sequence AGATCT, which is the recognition site for the restriction enzyme BglII. This enzyme cuts once in this segment of the CB4856 sequence and not at all in N2. Thus if we were to amplify this region from N2 and CB4856 worms using PCR and cut the PCR product with BglII, CB4856 would produce a doublet of about 500 bp each, whereas N2 would run as a single band of 1,000 bp. The other enzymes listed as distinguishing this polymorphism (e.g., MnlI and MboI) although technically correct, are not of much practical use, as they cut many times in both N2 and CB4856 sequences. Therefore, discerning these two largely identical digestion patterns (using a standard agarose gel) would be difficult or impossible.
Moving down to the unconfirmed SNP just below B0403, we find C36B7:21571 S=CT. The presence of a C in N2, and an A in CB4856, leads to the creation of a new site for the enzyme ApoI (consensus RAATTY; where R is an A or G and Y is a C or T. For a complete listing of abbreviations, see the back of the NEB catalog). Here we see that ApoI cuts five times in strain CB4856 (59, 405, 500, 638, 648). Directly above this, we see that the N2 digest is listed as “none”. Beware: this does not mean that CB4856 cuts five times with ApoI and not at all in N2! In fact, N2 cuts four times with ApoI (59, 405, 638, 648), just not at the middle position where the actual SNP is located (500). This is obviously misleading. By “none”, they just mean that the polymorphism results in no new enzyme sites that specifically cut the N2 sequence. Another thing to be aware of is that for non-palindromic sites, it may be the bottom (non-scripted) strand of DNA that is relevant.
Because many of the listed SNPs are not experimentally confirmed, the question arises: how many SNPs are actually real and is it possible to intuitively distinguish the real ones from the false ones? (The false ones are simply due to errors in the single sequencing reads of CB4856). For all non-confirmed SNPs, a probability index (Psnp) is given at the top of the page that contains the sequence information. For C36B7:21571, the Psnp is 0.9427, meaning that there is supposedly a 94% chance that the SNP is real based on the quality of the read. For a non-confirmed SNP, this is as good as it gets. In contrast, it is our experience that SNPs with Psnp indices below 0.5 are invariably bogus. Also note that non-confirmed nucleotide substitutions can now be cross referenced using the newer SNP database, described below. In addition to low-scoring substitutions, SNPs that result in single base-pair deletions or insertions within a run of repetitive nucleotides (e.g., A7 versus A8) are often suspect. Although some of these may turn out to be real, common sense dictates that sequencing errors are more likely to occur when attempting to distinguish between these sorts of differences than when comparing sequences such as ATG and ACG. Thus, you will want to use some discretion in your true/false predictions beyond the Psnp index. Of course, you will always want to substantiate any unconfirmed SNP before attempting any significant mapping exercises, no matter what the probability index or your intuition tells you.
The second, more recently developed database, can be accessed at: http://genome.ucsc.edu/cgi-bin/hgTables?org=C.+elegans&db=ce4&hgsid=109019391&hgta_doMainPage=1.
The primary advantage to this database is that, as mentioned above, it is based on the complete sequence of CB4856, and thus in theory should identify all known SNPs. However, as of this writing, the database identifies only nucleotide substitutions, but not small deletions and insertions. Given that this latter class comprises a substantial proportion of the differences between N2 and CB4856, this database is currently incomplete; the site developers are aware of this deficiency and a fix should be available in the near future.
From the page accessed by the above link, under the pulldown menus for the “group” and “track” inputs, select “Custom Tracks” and “cb4856_snps”, respectively. Use the defaults for all other categories. Under the “region” heading, select “position” and enter a specific chromosomal nucleotide location range, e.g., chrIV:500000-550000. Note that specific nucleotide numbers corresponding to any region of interest can be obtained from Wormbase. For example, entering the cosmid C32F10 on Wormbase and carrying out a “clone” search “ reveals the genomic location of this comsid to be “1:5,804,218…5,834,319”, which would be entered into the position box on the SNP site as chrI:5804218-5834319.
For the output section, several formats are available. For example “All fields from selected table” tabulates the changes and positions of the SNPs for that region. This output also provides a score of 40–63 for each SNP, where higher numbers indicate greater reliability. On average the database contains a false positive rate of ∼5%. Also very useful is the “sequence” option. This takes you to new page where you can enter the number of nucleotides on either side of the SNP that you would like displayed. For example, entering “50” into both the upstream and downstream boxes and clicking on “get sequence” will produce a list of SNPs, each displaying 101 nucleotide sequences (50 bp per line). In this case, the location of the actual SNP will be at position 51, or the first nucleotide on the second line. Note that the sequence displayed is always the N2 sequence, however, the specific change is indicated above the sequence. Thus, C/T would indicate that the nucleotide at position 51 is a “C” in N2 and a “T” in CB4856. These types of sequences can then be readily pasted into standard DNA analysis software to detect changes in RFLP patterns.
SNPs can also be accessed directly through WormBase, although somewhat less information is currently provided than the SNP-specific websites. To view these, simply go to your region of interest using the WormBase genome browser and select a reasonably-sized region (e.g., 20 kbp) for viewing under the “Scroll/Zoom” pulldown menu. Next, check the “SNPs” box towards the bottom of the page under “Variation Tracks” and click “Update Image”. This will display the predicted SNPs in the region as green or yellow diamonds, indicating RFLP and non-RFLP SNPs, respectively. In addition, SNPs that have been validated by additional sequencing or RFLP analysis are indicated. Clicking on the diamonds or adjacent text brings you to a new page where you have the option of viewing an expanded region (500 bp) surrounding the SNP. Alternatively, you can access SNPs through WormBase via: http://www.wormbase.org/db/searches/strains. Enter landmarks as directed and select “None” under the top Loci heading, “SNPs” under the middle option, and “All” under the bottom SNPs heading to view all verified and predicted SNPs in the region. Note that WormBase does not currently include reliability scores for predicted SNPs and there are no options for viewing different amounts of surrounding sequences or for identifying relevant restriction endonucleases. Nevertheless, the graphical interface is very straightforward and highly useful for visualizing the locations of SNPs within a small region.
With the consortium's sequencing of CB4856 and the expected future improvements of the databases, individual investigator's efforts to detect novel SNPs through sequencing relevant regions of CB4856 will likely be unnecessary in the very near future. Nevertheless, this can be accomplished by amplifying random intergenic sequences in one's region of interest from CB4856. In the past, we have usually amplified an ∼1,600-bp region from CB4856 and used two internal sequencing primers. More often than not, one will find at least a single difference within a region of this size.
An assessment of any SNP requires that the chromosomal region containing the polymorphism be amplified from worm genomic DNA (see below SNP PCR procedure). For confirmed SNPs from the original database, suggested primer sequences for amplification are indicated at the top of the window that contains the DNA sequence as well as by lower-case letters within the sequence text itself. These may often be a wise choice, although you will want to ultimately pick your primer sites based on two criteria: 1) the sites should enable you to make a clear distinction between N2 and CB4856 sequences, and 2) the primers should not anneal to other sites within the C. elegans genome. For the former concern, keep in mind that it is easier to distinguish the difference between one 800- and two 400-bp bands than one 400- and two 200-bp bands. This is because the smaller the bands get, the harder it may be to resolve subtle differences, and also the more likely that these bands will be partially obscured by the fuzzy (primer) bands that run near the bottom of many gels. Thus although it might be marginally easier to amplify the 400-bp band, the clouded interpretation will negate any positive benefits. In addition, it is extremely prudent to carry out a BLAST search on any primer that you intend to use prior to ordering. In the case that you uncover many perfect or near-perfect matches to the primer elsewhere in the genome (particularly to the 3′-most 15 or so nucleotides), go back to the drawing board and find another sequence that won't be as likely to give you high backgrounds.
Non-RFLP SNPs, although obviously harder to use than RFLP SNPs, can nevertheless be invaluable tools. This is particularly true once one has significantly narrowed down the genetic region containing the mutation of interest. At this point, you will have probably whittled down your informative recombinants to a workable number. Thus, any sequencing efforts will be less arduous and less expensive. Note that when testing non-RFLP SNPs by sequencing, always use an internal sequencing primer (not one of the outer primers used for amplifying the DNA), and place the 3′ end of the primer at least 50 base pairs away from the SNP site to avoid messy or ambiguous reads. In addition to sequencing, we have tested the use of the Surveyor(r) mutation detection kit from Transgenomic (tip of the hat to E. Haag). The detection method exploits the production of a small bubble of single-stranded DNA in re-annealed hybrids of N2 and CB4856 DNAs that contain sequence variations. A nuclease that recognizes the DNA distortion then cleaves both strands, which can be detected on a gel. Although this method is not nearly as straightforward as a restriction enzyme digest, it may avoid the time and expense of sending out PCR products for sequencing and could ultimately make non-RFLP SNPs more attractive to use.
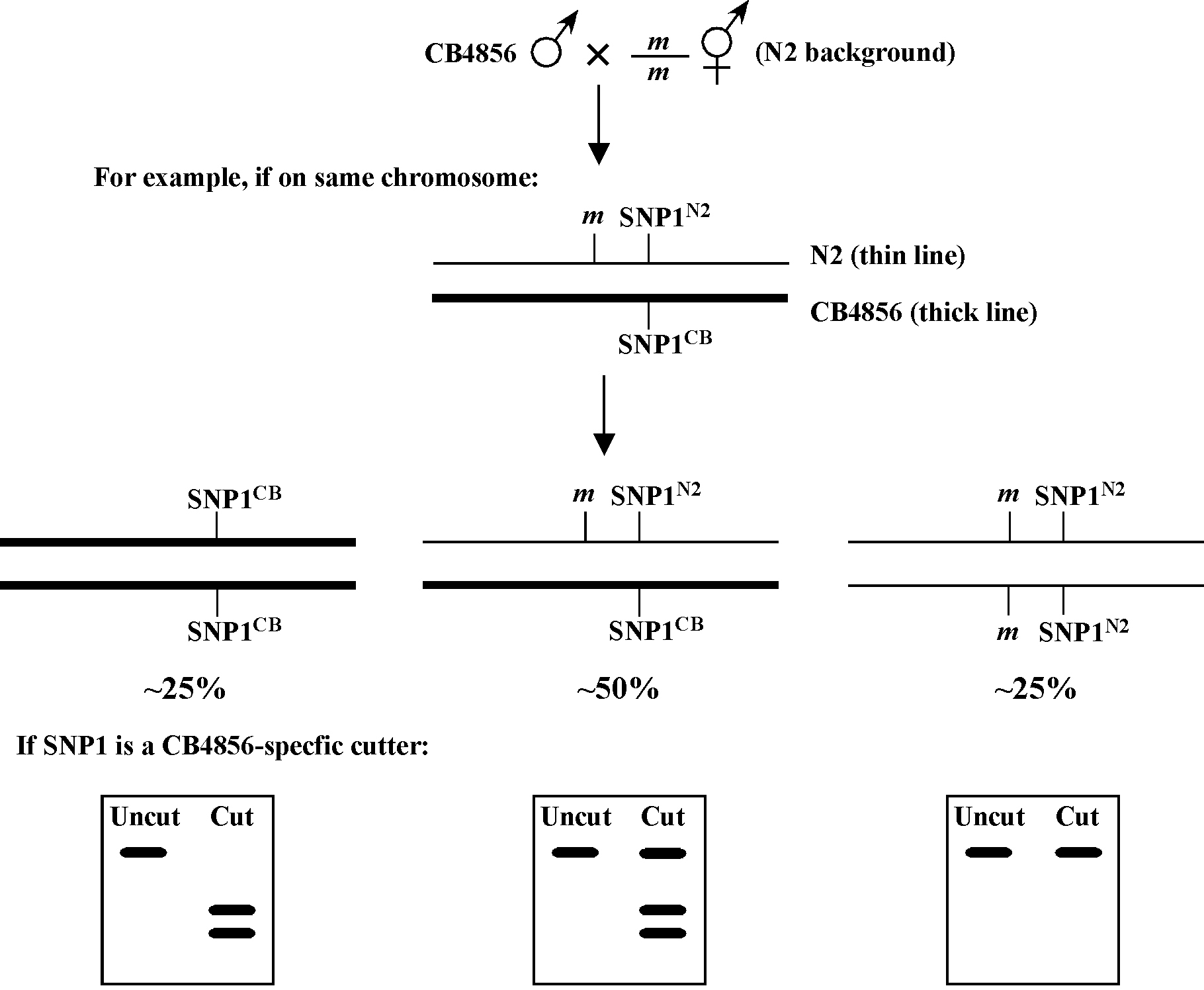
For all standard SNP mapping, you will want to generate and maintain a stock of CB4856 males. The males are then crossed into your mutant strain to generate heterozygous cross progeny that are allowed to self, leading to the regeneration of your homozygous mutant. In certain situations (such as for mapping suppressors and enhancers), you may want to generate versions of your mutant strains that have been extensively outcrossed to CB4856, and then use males from these stocks instead of CB4856 (also see other chapters). Figure 1 shows one basic scheme for 2-point SNP mapping. Just as in 2-point genetic mapping, the closer your mutation lies to the given SNP being tested, the less likely that a homozygous mutant will harbor a CB4856 allele of that polymorphism, and the more likely that there will be a significant over-representation of N2 homozygous loci among mutant animals. In fact, if the SNP you are testing lies very close to your mutation, you may observe nearly 100% of mutant animals to be homozygous for the N2 locus at that SNP.
In the event that your mutation lies on the far arm of a chromosome, linkage can still be reliably detected using a central SNP. For example, if the mutation and SNP are separated by 25 map units, ∼56% of homozygous mutant animals should be homozygous N2 for the SNP (38% will be N2/CB and 6% CB/CB). In the case that your mutation is unlinked from the SNP being tested, homozygous mutants will segregate N2/N2: N2/CB : CB/CB animals in the standard 1:2:1 ratio. Thus 75% of the homozygous mutant animals will be either N2/CB (50%) or CB/CB (25%), whereas only 25% will be N2/N2. For additional details on calculating predicted percentages based on genetic distance see Two-point mapping with genetic markers.
One helpful tactic when mapping viable homozygous mutants is to use CB4856-specific cutters as SNPs whenever possible. The reason for this has to do with making the interpretation of data from animals that are potentially a mixture of N2 and CB4856 (50% in the case of non-linkage) as unambiguous as possible. Namely, if you were to use an N2-specific cutter and observed a small amount of residual uncut PCR product on your gel (as well as bands indicating cleavage), you might conclude that the uncut DNA demonstrates the presence of some CB4856 DNA at that locus in that isolated strain. Thus, the re-isolated mutant strain would be scored as a mixture of N2 and CB4856 (N2/CB). However, cleavage by restriction enzymes can be variable and is often incomplete, and therefore you couldn't be certain that the residual uncut band was due to the presence of non-cleavable (CB4856) DNA or simply the result of an incomplete digest or possibly even a weak coincident background band from the PCR. Contrast this with using a CB4856-specific cutter: in this situation, the appearance of even a small amount of cut DNA of the correct sizes would strongly support the presence of some CB4856 DNA at that locus. Of course, it is also true that homozygous CB4856 DNA may not cut to completion either. However, the distinction between N2/CB and CB/CB classes of strains is far less critical than recognizing strains that are truly homozygous for N2. Thus, the assignment of N2 homozygous and N2-non-homozygous isolates is much cleaner when using CB4856-specific cutters in this scenario.
The above logic may, however, be reversed when mapping homozygous nonviable mutants such as those that result in embryonic or larval lethality. In this situation, you obviously won't have the option of propagating a homozygous mutant strain. In theory, one could carry out PCR on individual arrested larvae or embryos, however, this can be technically challenging and won't provide you with stable strains for the purpose of any further mapping refinements (see below). An alternative scheme is shown in Figure 2.
Here, instead of re-isolating homozygous mutants that are N2/N2 at the relevant loci, you will want to ultimately select for animals that are CB/CB at this loci by identifying strains that fail to segregate your mutant phenotype. The same relative ratios as described above will apply, although the expectation is that SNPs residing close to your mutation of interest will be approaching 100% CB/CB, whereas unlinked SNPs will be 25% N2/N2, 50% N2/CB, and only 25% CB/CB. Also, note that the logic of using CB-specific cutters is also reversed, such that you will want to use N2 specific cutters when using this approach.
There are several other considerations in carrying out two-point SNP mapping. First, because we typically pick only a limited number of animals into one tube for any given PCR reaction, the possibility of randomly picking several animals that are homozygous for a given SNP from a mixed plate increases as we pick fewer animals. For this reason, it is always advisable to pick at least five adults for each tube, or to pick piles of larvae from starved plates. Alternatively, one can rapidly prepare DNA stocks from whole plates using standard protocols, thereby providing a source of DNA for multiple PCR reactions. Also, when propagating strains, make sure either to chunk the plates (by transferring a piece of the agar) or to transfer large numbers of animals by pick to preserve the heterogeneous status of the mixed plates. In addition, it is critical to limit the number of generations that the strains have undergone prior to SNP testing. Part of the rationale for this is described above. Moreover, it has been our experience (and that of others) that mixed populations may drift towards homozygosity of specific N2 or CB4856 alleles or chromosomes not simply via a random process but through active selection. For example, we have observed a strong selection in N2/CB strains to homozygose the N2 X chromosome. Thus it is essential to obtain the required SNP data from your collection of mutant isolates rapidly to avoid the biasing that may take place after multiple generations. Also, it is a better idea to let your plates starve out while you're doing the testing than to continually passage them to new plates, thus minimizing any selection that may (want to) take place.
Once you have assigned a linkage group to your mutation based on 2-point SNP mapping data, it may be possible to refine the position of the mutation by testing other regional SNPs. For example, if you had assigned your mutation to a specific chromosome based on the observation that 35/40 of the mutant (m/m) isolates were homozygous for the N2 locus, it should be possible to test new SNPs that are several map units to either side. Thus you may find a new locus where 39/40 mutants contain the homozygous N2 allele, indicating a closer linkage to your mutation. You may also want to choose a good pair of genetic markers for quickly confirming your SNP result by standard 2-point mapping. This will not only serve the purpose of independently confirming your SNP results but will also allow you to begin the process of genetic three-point mapping (see Three-point mapping with genetic markers).
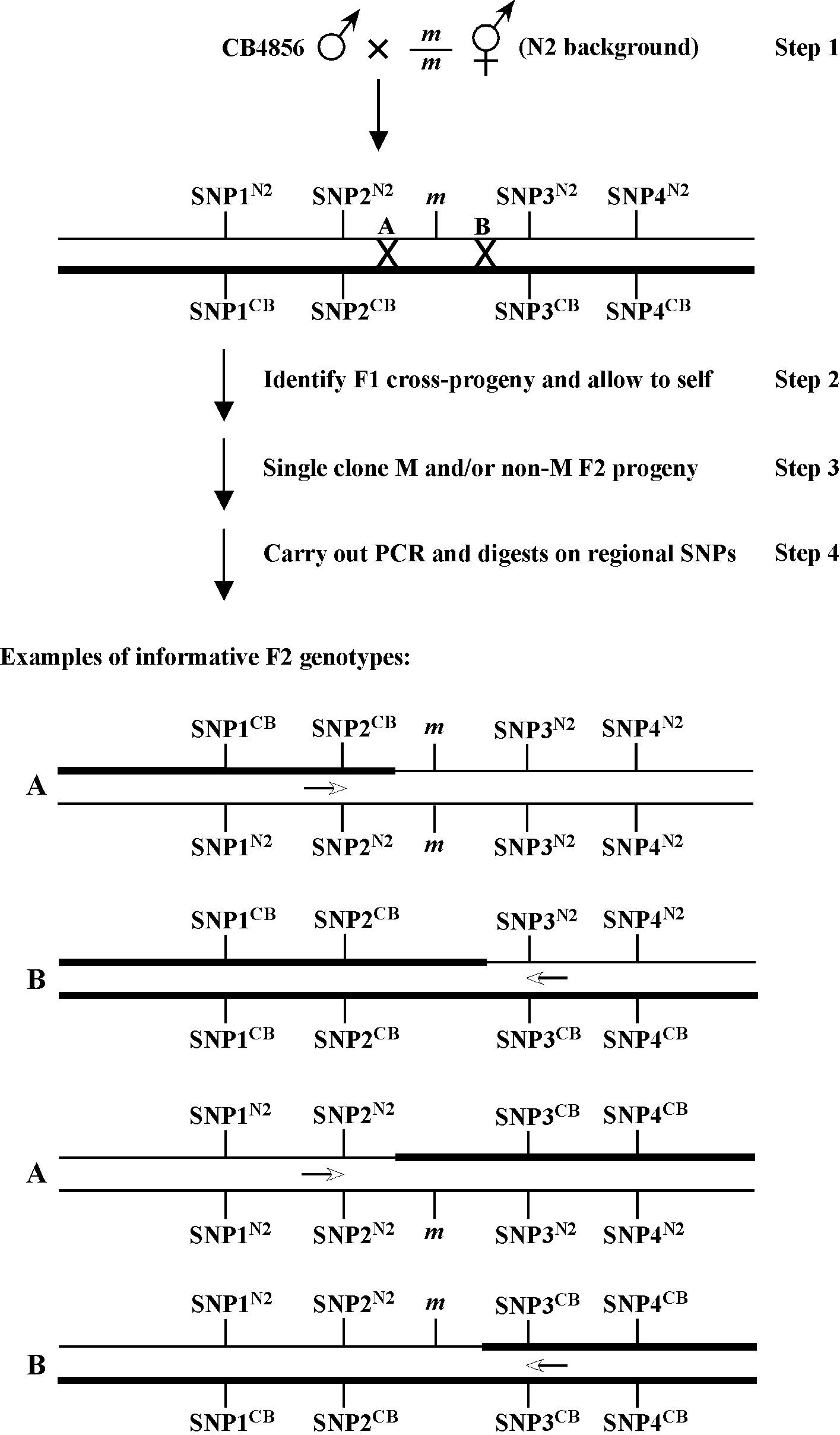
In the not too distant future, it will likely be possible to rapidly and economically identify mutations of interest using high-throughput sequencing approaches, tiling arrays, or other methods that can reliably detect single-base-pair alterations. However, even using these types of approaches, it will be beneficial to limit the region of interest to a relatively small genomic segment (e.g., several mega bases or less) in order to carry out more focused analyses. 2-point SNP mapping provides a means to accomplish this through the ability to define molecular endpoints. A generic scheme for doing this is shown in Figure 3. Unlike the non-recombinant chromosomes shown in Figures 1 and 2, potential F2 genotypes are depicted in which random recombination events have taken place either to the immediate left (A) or right (B) of the mutation of interest (m). For this approach to be effective at least 100–200 F2 animals must be cloned and allowed to self. Following examination of the F3 generation for the presence or absence of M animals, a linear series of SNPs (e.g., 1–4) can be assayed to identify those strains in which a recombination event took place in an interval proximal to your mutation of interest.
Figure 3 shows the genotypes of several classes of informative F2 recombinants. In the case of the genotype shown at the top, SNP2 can be reasonably established as a solid left endpoint. This is because m, a recessive mutation, is homozyogous, even though the strain is heterozygous for CB4856 DNA to the left of SNP2. In the second example from the top, SNP3 can likewise be established as a right endpoint. This is because such an animal will never throw M (m/m) animals, despite the presence of N2 DNA to the right of SNP3. The lower two genotypes represent heterozygous m/+ animals that throw one-quarter M progeny. In both cases, the presence of homozygous N2 or CB4856 sequences on this chromosome further enforce SNP2 and SNP3 as boundary endpoints. This kind of approach can be effectively used to implicate regions of several map units or less in a relatively short time frame. Nevertheless, given that recombination events within ones region of interest cannot be selected for using this approach (as in 3-point genetic and SNP mapping), relatively large numbers of F2s must be examined in order to identify relevant genotypes. It is worth noting, however, that such endpoint determinations would not be possible through traditional 2-point mapping using standard genetic markers.
10% PCR Buffer
| 100mM Tris-HCl (pH 8.8) |
| 500mM KCl |
| 0.8% NonIdet p40 detergent |
Lysis Buffer
| 50mM KCl |
| 10mM Tris-HCl (pH 8.3) |
| 2.5mM MgCl2 |
| 0.45% Nonidet P-40 |
| 0.45% Tween 20 |
| 0.01% (w/v) gelatin |
| (Autoclave and store aliquots at 20°C) |
Proteinase K (10.0 mg/ml)
MgCl2 (25mM)
dNTP's (2.5mM each)
DNA polymerase (5 units/Rxn)
PCR primers (10.0 pmol/ml)
Add 1 μL of 10.0 mg/mL proteinase-K to 99 μL of lysis buffer and mix well. It may be necessary to scale up depending on the number of PCR reactions to be done. Add 3.0 μL of mix to the open lids of several 0.2 ml PCR tubes. Pick 1 to 5 worms and place them in the drop of solution in the tube lid by swirling pick within the liquid. Be sure that all the worms are in the lid before proceeding. Next, close the lid of the tube and spin in a microfuge for 10 seconds. You can transfer worms to multiple tubes prior to spinning, provided the samples are not left at room temperature for too long. After centrifugation, place the tubes on ice in a 96-hole PCR tube rack. When finished, place the rack at −70°C for 45 minutes (or longer). Next, place the tubes in a thermocycler and run the following sequence: 65°C 1 hr, 95°C for 30 min., 4.0°C hold. The proteinase-K is active at 65°C but is efficiently de-activated at 95°C. The heat-deactivation is crucial because the proteinase can cleave the polymerase during PCR.
After the lysis reaction is completed, the lysate can be used directly for PCR or stored at −70°C. Prepare a master mix containing the following ratios of ingredients:
| 13.5 μL dH20 |
| 2.5 μL 10x PCR buffer |
| 1.5 μL 25.0 mM MgCl2 |
| 1.25 μL forward primer |
| 1.25 μL reverse primer |
| 1.0 μL dNTP's (2.5 mM each) |
| 1.0 μL Taq DNA polymerase (∼ 5U) |
Mix the solution well and add 22.0 μL to each tube containing 3.0 μL of worm lysate. Be aware that the ingredients for the PCR master mix can vary depending on the polymerase used. Read the manufacturer's recommendations carefully. Most manufacturers provide 10x buffer and a tube of MgCl2 or MgSO4 with the polymerase. Also, when putting together the master-mix, don't forget to make a bit extra for the PCR fairies. For example, if you are carrying out 20 reactions, make enough mix for 22.
It is often difficult to guess at ideal thermocycling conditions for PCR (principally the annealing temperature). Therefore, it is best to determine these conditions prior to conducting PCR for mapping purposes. If you have access to a thermocycler that is capable of running temperature gradients, ideal conditions can be determined in a single step. Set the annealing temperature for the center rows of the machine at the Tm given on the lyophilized primer tubes provided by the manufacturer. Next, program the machine to run a 10°C temperature gradient. This often means that during the annealing step, the leftmost column will be 10°C cooler than the center columns and the rightmost column 10°C warmer. This effectively results in a 20°C difference between the left and right columns. Most gradient thermocyclers allow you to view the exact annealing temperature in each column for a given reaction. Record these so you can keep track of the ideal annealing temp for future reactions. Prepare 12 reactions for N2 and 12 for CB4856. Place them in parallel rows within the machine (i.e. A1 - A12: N2 and B1 - B12: CB4856). For the initial PCR reaction, set the following conditions:
| 1. 95°C - 2.0 min. (initial denaturation) |
| 2. 95°C - 45 sec. (denaturation) |
| 3. Annealing step - 30 sec. (see above) |
| 4. 72°C - 2.0 min/kB (extension) |
| 5. Repeat steps 2, 3, and 4, 30 to 40X |
| 6. 4.0°C hold |
After the reaction is finished, run a 0.8–1.2% agarose gel and stain with EtBr. The ideal annealing temp will correspond to the lane/lanes on the gel where a single strong PCR product of the correct size is seen. This is usually the same temp for N2 and for CB4856. Aberrant bands can often be attributed to non-specific annealing of the primers to other genomic fragments. This can usually be corrected by using a higher annealing temperature. The gradient reaction is valuable for empirical determination of annealing temperature in one step. However, if a gradient machine is not available, set the initial annealing temp at or somewhat above (5–10°C) the Tm on the primer tubes.
Once the ideal annealing temp is found, digest both the N2 and CB4856 samples with the appropriate restriction enzyme to be sure that the difference in fragment sizes correspond to the SNP database predictions and can be readily determined. Usually we digest ∼5 ml of the PCR reaction in a total volume of 10 ml for several hours. It is often useful to run the un-digested PCR product next to digested product for each recombinant. This is particularly helpful when primer sets or conditions produce non-specific amplimers that might be confused with digest products. Also, don't forget to always include N2 and CB4856 control reactions when doing SNP mapping experiments. For this purpose, we usually have on hand genomic DNA from large-scale preps of these two strains.
We would also mention that primer design is of the utmost importance in getting SNPs to work. A number of free WEB-based programs are available that will aid in minimizing secondary structure and optimizing and matching melting temperatures. We also find it useful to conduct BLAST searchers of the proposed primers to avoid those that may be complementary to multiple locations within the genome. In some instances, bad primer sets cannot be predicted. In these cases we simply order two more primers and then perform a mix and match experiment with the old and new primers to determine the pair that works the best. We also prefer, when possible, to use primers that amplify fragments of 750–1,000 bp. This is somewhat larger than many of the suggested primers sites often described on the SNP database (∼500 bp). In our experience, slightly larger fragments are somewhat easier to interpret, since the cut products still run well above the junk (primer-dimer bands) at the bottom of the gel. However, in some cases, this advantage isn't necessary or may be offset by the decreased efficiency of amplifying a larger band.
Jakubowski, J., and Kornfeld, K. (1999). A local, high-density, single-nucleotide polymorphism map used to clone Caenorhabditis elegans cdf-1. Genetics 2, 743–752. Abstract
Koch, R., van Luenen, H.G., van der Horst, M., Thijssen, K.L., and Plasterk, R.H. (2000). Single nucleotide polymorphisms in wild isolates of Caenorhabditis elegans. Genome Res. 11, 1690–1696. Abstract Article
Swan, K.A., Curtis, D.E., McKusick, K.B., Voinov, A.V., Mapa, F.A., and Cancilla, M.R. (2002). High-throughput gene mapping in Caenorhabditis elegans. Genome Res. 7, 1100–1105. Abstract Article
*Last revised June 30, 2008. Published September 25, 2008. This chapter should be cited as: Fay, D. and Bender, A. SNPs: Introduction and two-point mapping (September 25, 2008), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.93.2, http://www.wormbook.org.
Copyright: © 2008 David Fay and Aaron Bender. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: davidfay@uwyo.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.