Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergDominant alleles were first described by Mendel to account for the patterns he observed with respect to flower color. For example, red flowers are produced when the R allele is present in one or more copies (i.e., genotypes R/r and R/R), versus white flowers, which are produced only when the r allele is homozygous (r/r). In this case, the R allele is said to be dominant to the r allele, which is recessive. As the study of genetics has matured, the common definition of dominance has come to refer to alleles whose phenotype is manifest when present as a heterozygote (R/r). Dominant alleles may also be phenotypically deterministic when present along with two or more recessive alleles (e.g., R/r/r), a situation sometimes encountered in transgenic strains or with free duplications.
A good example of a dominant allele in C. elegans is the rol-6(su1006) allele, which causes a roller (Rol) phenotype. rol-6(su1006) animals exhibit the Rol phenotype when they are of the following genotypes: rol-6/rol-6; rol-6/+; or +/rol-6. Because the Rol phenotype is observed when a single mutant copy of rol-6(su10060) is present, the rol-6(su1006) allele is said to be dominant. Keep in mind that not all alleles of a particular gene will be dominant; there are several rol-6 alleles that exhibit recessive phenotypes. Dominance or recessivity are allele-specific properties. They are not gene-specific properties.
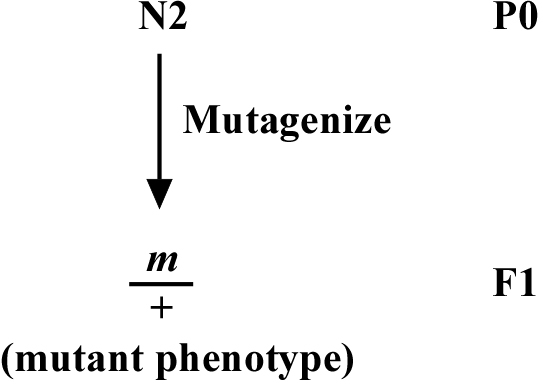
Depending on the particular developmental question in which you are interested, the systematic isolation of dominant alleles may be desirable. If you decide this is the case, the isolation of dominant alleles is straightforward. Although the typical genetic screen in C. elegans often aims to isolate recessive, loss-of-function alleles, as shown in Figure 1, the isolation of a dominant mutation requires one to simply screen the F1 generation (i.e., the self-progeny of mutagenized P0 worms), as shown in Figure 2.
This is trivial. One can also isolate dominant alleles in the first screen, because dominant mutations will exhibit the mutant phenotype in the F2 generation as well as the F1 generation. In a non-clonal screen, where F2 worms on a plate will derive from several different P0 animals, one may not notice that a particular allele is dominant until outcrossing fails to eliminate the mutant phenotype in cross-progeny. When outcrossing any newly isolated mutation, one should carefully observe the genetic behavior of an allele to determine whether it is dominant or recessive.
Whether your dominant mutation was isolated on purpose or by chance, the next step will be to map it to a chromosome. You'll recall from earlier sections that recessive alleles are initially crossed to wild type and then progeny of the heterozygous animals are then scored for segregation of the mutations and known genetic markers. If an allele is dominant, however, it is necessary to change our thinking slightly when scoring the segregation of phenotypes in mapping strains. Because we cannot determine whether a phenotypically mutant animal is heterozygous or homozygous for the dominant allele by simple observation, we use the alternative strategy of mapping the absence of our dominant allele. Once one has thought about it carefully, it can often be easier to map true dominant mutations than recessive mutations.
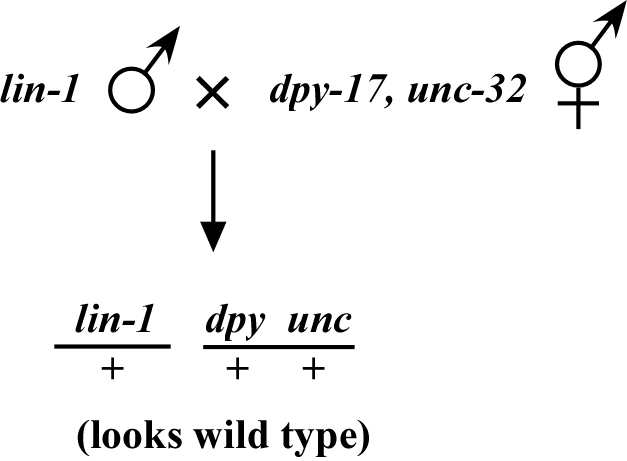
The phenotype of a recessive mutation disappears when crossed into a mapping strain. Consider lin-1, which causes a multivulval (Muv) phenotype when crossed into a strain as shown in Figure 3. We can then score the co-segregation of lin-1 and dpy-17 unc-32 in the normal manner by picking Muv non-Dpy Uncs and noting how often the Dpy Unc phenotypes co-segregate with lin-1Muv.
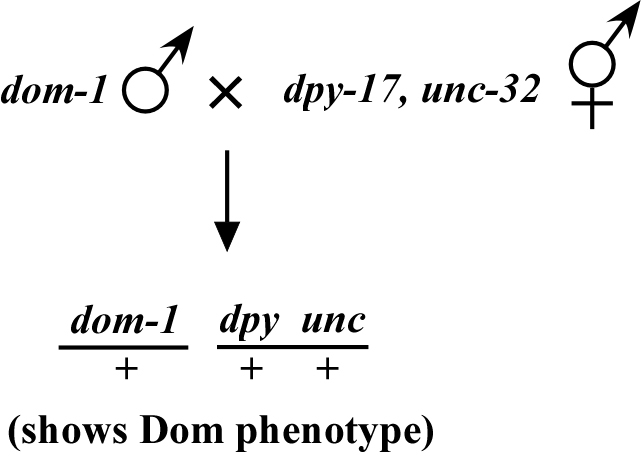
Now consider a dominant mutation that, in contrast to lin-1, exhibits the mutant phenotype when the allele is present as a heterozygote. When we cross into our chromosomal mapping strains, all the heterozygous cross progeny will exhibit the mutant phenotype. Let's consider an imaginary dominant mutation, dom-1, as shown in Figure 4, which we'll say causes a spiked-head phenotype. These dom-1/+ heterozygotes will display the spiked-head phenotype and will be indistinguishable from dom-1 homozygotes. Thus, we will be unable to score the segregation of dom-1 with dpy-17, unc-32 by following spiked-head animals because we won't know whether the animals are dom-1/+ or dom-1/dom-1.
The trick to mapping such true dominant mutations is to follow the animals that do not display the dominant phenotype. In other words, ignore the dom-1 phenotype and look only for those animals that are wild type. The reason for this is as follows: as we can always tell when dom-1 is present because of the dominant spiked-head phenotype, we follow the absence of dom-1 and note how often the markers segregate with non-spiked-head animals.
The results from chromosomal mapping of dominant mutations are apparent in the F2 generation. Because you are following the absence of dom-1, at this stage you are looking for non-spiked-head animals. If dom-1 is on the same chromosome as your markers and is relatively close (as shown in Figure 5; case #1), then the only animals with normal heads will be the Dpy Uncs. Also if you were to pick spiked-head animals and look for those that throw 100% spiked-head animals in their progeny (dom-1/dom-1), few or none will throw Dpy Uncs. If, however, dom-1 is on a different chromosome from the markers (as shown in Figure 5; case #2), then it will segregate independently from the markers, and 3/4 of the Dpy Unc progeny will display spiked heads. Also in this case, 2/3 of the spiked-head (non-Dpy Unc) progeny that throw exclusively spiked-head progeny (dom-1/dom-1) will throw Dpy Uncs.
Once we have our dominant mutation mapped to a chromosome, it is similarly easy to collect data for three-point mapping. We'll start again with our balanced strain that is heterozygous for both dom-1 and our markers. This strain will have the spiked-head phenotype. In this example, we'll assume dom-1 lies between dpy-17 and unc-32. When looking for recombinant Unc-non-Dpy or Dpy-non-Unc animals, we will know immediately whether or not the recombinant picked up the mutant dom-1 allele because of its dominance. Recombinants that pick up the dom-1 allele will have a spiked head, and recombinants that don't pick it up will have wild-type heads. Quick and easy.
Why do some mutations act in a dominant fashion? Below we examine some different mechanisms through which a mutation can confer a dominant phenotype. In certain situations, different dominant alleles may require different mapping strategies. These situations must be managed on a case-by-case basis. In each example below, we will consider the fictional dom-1 gene and imagine different situations that could give rise to various types of dominant alleles in dom-1.
This describes a situation in which one copy (haplo) of a wild-type gene is not enough to provide wild-type function when the other copy is compromised. This can only occur for loss-of-function alleles. Consider again our fictional dominant mutation, dom-1. Let's assume that a certain threshold of dom-1 activity is required to avoid the abnormal spiked-head phenotype; two copies of the wild-type gene are required to achieve that threshold, and any drop below that threshold allows the mutant spiked head to form. Mutations in dom-1 that reduce or eliminate its activity would therefore behave dominantly because in heterozygous animals, the single remaining wild-type copy of the dom-1 gene would be insufficient to provide the wild-type levels of gene activity. Thus, the loss-of-function dom-1 mutant allele may produce a similar phenotype whether present in one or two copies and behaves in a dominant fashion. Alternatively, dom-1/+ heterozygous animals may display a phenotype that is quantitatively or qualitatively different from homozygous dom-1/dom-1 animals, since the former would still retain half the normal gene dose.
These typically occur when the mutant allele does not function normally and either directly inhibits the activity of the wild-type protein (usually through dimerization) or inhibits the activity of another protein that is required for the normal function of the wild-type protein (such as an activator or downstream component in a pathway). Although this situation can effectively result in the loss of function of the wild-type protein, it differs markedly from haploinsufficiency. Consider an animal that is heterozygous for a dominant-negative allele of dom-1. In this case, we'll also imagine that the single wild-type copy of dom-1 would normally provide enough dom-1 activity to avoid the spiked-head phenotype. However, because of a dominant-negative version of dom-1 would actually interfere with the function of wild-type dom-1 its activity is further reduced and a mutant phenotype results.
A well-known example of a gene that can incur dominant-negative mutations is the small GTPase Ras. These dominant-negative alleles of Ras are not functional themselves because they preferentially bind GDP and stay locked in the inactive state. In addition, they also prevent the Ras exchange factor (which binds Ras-GDP and catalyzes GDP/GTP exchange and subsequent Ras activation) from acting on wild-type Ras, essentially killing all Ras activity.
Also termed hypermorphs, these refer to mutations that result in elevated levels of gene activity. In some cases, dominant GOF mutations may acquire novel biochemical activities, in which case they may be referred to as neomorphs. It is possible to imagine numerous scenarios that might lead to the removal of normal regulatory constraints and the enhancement of protein activity. For example, a mutation in the promoter region could lead to overexpression of the gene and the saturation of negative regulatory pathways. Alternatively, point mutations in a region of a gene important for its regulation could lead to inappropriate activity and mutant phenotypes. Let's revisit dom-1 and imagine it is an enzyme whose activity promotes head development. Assume that normal levels of dom-1 activity result in normal head development and any dom-1 activity above normal levels results in a spiked head. Also assume that a negative regulatory phosphate group is added to an N-terminal serine when dom-1 activity gets to the threshold required for normal development. A point mutation that makes this serine phosphorylation impossible (e.g., Ser → Ala) could remove the negative regulation of dom-1 and allow its activity to proceed unchecked, thus leading to the spiked-head phenotype. In short, too much of a good thing can lead to developmental abnormalities.
It is actually quite typical for dominant alleles to behave in a partially dominant fashion. Alleles are designated semi-dominant when the penetrance of the phenotype in heterozygous animals (dom-1/+) is less than that observed for homozygous animals (dom-1/dom-1). For dom-1, this would be the case if dom-1/dom-1 animals were 100% spiked head and dom-1/+ animals were 60% spiked head. This is an important point, as the basic mapping strategies outlined above were assuming 100% dominance. In practice, this is not necessarily that difficult to deal with, as the presence of the mutation will always be seen by the next generation, as is the case for any recessive allele. Thus mapping a semi-dominant mutation will simply require following progeny for an extra generation to distinguish between dom-1/+ and +/+ animals.
To attempt to distinguish between various classes of dominant mutations, a number of genetic tests can be performed. For example, to determine if a mutant phenotype observed in a heterozygous animal is due to haploinsufficiency, one can directly examine animals that are heterozygous for a chromosomal deficiency that removes the entire gene (as well as a number of other genes presumably). Alternatively, if a deletion or null allele of the gene exists, placing this mutation over the wild-type chromosome could provide an even cleaner answer. In addition, to distinguish haploinsufficieny effects from hypermorphic mutations, one can further compare homozygous mutant animals (dom-1/dom-1) with animals that are heterozygous for the mutation and the deficiency (dom-1/Df). If the homozygous mutants show a more severe phenotype than the mutant allele over the deficiency, then it is likely that the mutation is at least partially dominant, although one can have both dominance and haploinsufficient effects for the same allele.
In addition, a hypermorphic mutation would be expected to exert an effect even in the presence of two normal copies of the gene. Thus, a genetic test of this can be carried out using a free duplication that contains a wild-type copy of the gene, which is examined in the background of the heterozygous mutant (e.g., dom-1/+; Dp). A further test is to examine dom-1/dom-1; Dp animals. In this case, if the mutant allele is not hypermorphic (only LOF associated with haploinsufficieny), the phenotype of this animal should be no more severe than dom-1/+ animals and may even be less severe if the dom-1 allele contains some residual activity.
Other questions may be more difficult to answer genetically, particularly in the absence of knowing or understanding the molecular functions of the gene. For example, distinguishing dominant negatives from dominant gain of function alleles may be difficult in a vacuum. The ability of RNAi to phenocopy or enhance a dominant mutation would suggest that the mutation is a dominant negative, although a negative result in this case is difficult to interpret. Also, if the gene is cloned, then attempts to overexpress the wild-type version of the gene product may be informative in this regard, as phenocopy would indicate a hypermorphic mutation. Also, a dominant negative might be expected to be less penetrant in a background that contains one or more copies of the wild-type gene (e.g., dom-1/dom-1 versus; dom-1/dom-1; Dp), although a number of hand-waving explanations can theoretically weaken these types of arguments.
In closing, we refer readers to a number of published papers dealing with various types of dominant mutations in C. elegans (see below). We hope you enjoyed this discussion of dominant alleles. Now get back to work, dammit (A. Spencer).
Greenwald, I.S., and Horvitz, H.R. (1982). Genetics 101, 211–225. Abstract
Han, M., and Sternberg, P.W. (1991). Genes Dev. 5, 2188–2198. Abstract
Han, M., Aroian, R.V., and Sternberg, P.W. (1990). Genetics 126, 899–913. Abstract
Mitenko, N.L., Eisner, J.R., Swiston, J.R., and Mains, P.E. (1997). Genetics 147, 1665–1674 Abstract
*Edited by Victor Ambros. Last revised May 6, 2005. Published February 17, 2006. This chapter should be cited as: Fay, D. and Spencer, A. Genetic mapping and manipulation: Chapter 8-Dominant mutations (February 17, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.97.1, http://www.wormbook.org.
Copyright: © 2006 David Fay and Andy Spencer. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: davidfay@uwyo.edu or aspencer@biochem.wisc.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.