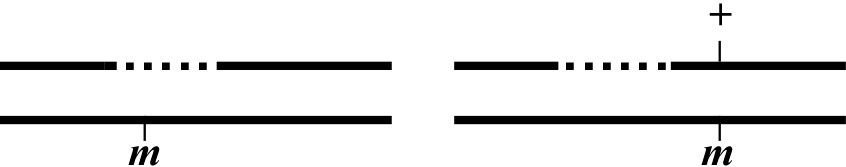Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergDeficiency (Df) mapping works great, when it works! Deficiencies refer to specific deleted regions within chromosomes. The sizes of Dfs vary greatly from just a few cosmids wide to the absence of a large portion of the chromosome. The endpoints of the Df may have been determined precisely using molecular techniques or may be rough guesses based on genetic tests with various mutations. Homozygous Df animals are almost always embryonic lethals, as removal of multiple genes usually includes some that are necessary during early development. One of the traditional strengths of Df mapping is that it can provide clear-cut irrefutable endpoints for ones region of interest (as opposed to the statistical arguments often associated with three-point mapping). The problem in mapping with Dfs is that although positive results are generally unambiguous, negative results can be more difficult to nail down. In addition, the availability of SNPs to define true endpoints has to some extent removed much of the allure of working with deficiencies. Nevertheless, Dfs are still extremely useful for certain types of genetic analyses, such as determining whether or not mutations are null (also see Dominant mutations). The basics of Df mapping strategy are shown in Figure 1.
In the case on the left, the deficiency (indicated by the dashed line) deletes the gene of interest and as a result fails to complement (does not rescue) the mutant allele on the opposite chromosome. Animals with such a genetic configuration will generally show the original mutant (M) phenotype. An exception to this can occur when the mutation is a hypomorph (partial loss of function). In this case, m/Df animals may show a new "M" phenotype that is more severe than the phenotype of m/m animals. In the case on the right, the mutation is outside the Df and the M phenotype will not be displayed. If the breakpoint of the Df is near m, one may have generated a well-balanced heterozygote, as recombination is often reduced in the immediate vicinity of the Df. Nevertheless, it is important to keep in mind that recombination might occur between the right breakpoint and m, thereby destroying the balanced stock.
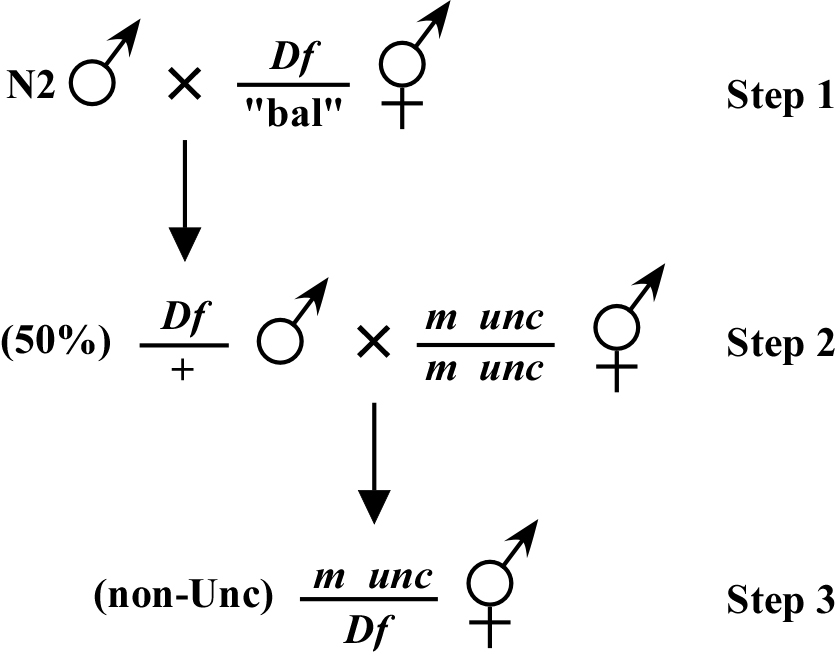
The way most deficiency mapping is done is as follows: You set up a situation where you are looking for a mutant phenotype in the F1 generation of the cross. For example, as shown in Figure 2, your mutation, which is linked to an unc or some other visible marker in cis, is crossed to male animals (step 2) that were created by mating N2 males into a balanced deficiency strain (step 1; provided Df is not on X). In this case, the unc mutation linked to your mutation is known to be outside this particular Df. In this scenario, if the mutation is within the Df, you will observe non-Unc animals displaying the M phenotype in the F1 generation (step 3). The presence of the linked unc marker is necessary here to clearly identify cross-progeny. For Df mapping, it is important to set up as many mating plates as possible (10-15 is not an excessive number) to guarantee generation and detection of the m/Df genotype. Failure to observe the M phenotype would indicate that m is outside the Df, provided that on most of your mating plates you observe good numbers of non-Unc cross-progeny. However, to ensure a correct interpretation of negative results, it is necessary to test whether animals of the genotype m/Df have a wild-type phenotype. Such an animal can be identified by the segregation of M and Df (usually embryonic lethal) phenotypes in the F1.
As an alternative approach, shown in Figure 3, you can also mate your mutation via the male into the Df strain (step 2) and look for your M phenotype in the first generation of cross-progeny (step 3). In this case, it is best to have the linked unc mutation (or other cis marker) inside the Df so that cross-progeny of the desired phenotype can easily be identified via the Unc phenotype. This approach can also be used without any linked marker where one just looks directly for the appearance of the M phenotype in the F1 generation. As with the previous example, if the result appears negative (the m is outside the Df), it is important to try and verify this by cloning supposed F1 cross-progeny and making sure that some of the phenotypically wild-type F1s throw progeny of both M and Df phenotypes.
Duplications (the Dps) are used less frequently than Dfs and are probably of less utility. Free duplications are autonomous pieces of DNA derived from normal chromosomes. They are usually relatively small as compared with full-length chromosomes and exhibit segregation properties that are independent of other chromosomes, including the chromosome from which they were derived. In many ways they most resemble extrachromosomal arrays and, like arrays, tend to be significantly less stable (especially meiotically) than normal chromosomes. Dps will vary significantly in their genetic stability, and some published information exists describing the properties of various Dps. Animals that contain a Dp will effectively be triploid for the genes that lie within the Dp. Dps are often used to balance a homozygous lethal mutation.
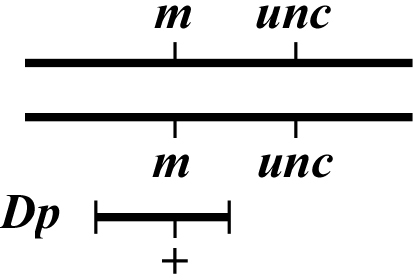
The idea in mapping with free Dps is to determine whether or not the mutation of interest lies within the duplicated region. If it does, then one would observe "rescue" of the mutant phenotype. To do this, one will set up crosses that ultimately lead to the isolation of the genotype shown in Figure 4. This can in theory be accomplished using several approaches but usually takes a number of steps as one must re-homozygose animals for m. Having a marker linked to (m) but outside the region covered by the Dp can be useful for identifying candidate (m/m) animals. As with Dfs, it will generally be quite obvious when one's mutation lies within the Dp but rather more difficult to prove that it definitely lies outside. Creating a chromosome where the mutation is flanked by two visible markers (one outside and one inside the Dp) can help to clarify this issue.
One phenomenon to be aware of if you are introducing a free Dp into your genetic background via a male concerns the behavior of some free Dps during male meiosis. Namely, certain Dps (including some derived from the autosomes) tend to pair and subsequently disjoin from the lone X chromosome, leading to sperm that contain either the X or the Dp, but not both (Herman et al., 1979). The functional consequence of this is that the majority of Dp-containing cross progeny tend to be males, which may not be particularly useful if the next step in your strategy is to self the Dp-containing hermaphrodites. Thus, it may be prudent to set up additional mating plates and pick more potential Dp-containing hermaphrodites in these situations to ensure that sufficient hermaphrodites containing the Dp are obtained.
Herman, R.K., Madl, J.E., and Kari, C.K. (1979). Duplications in Caenorhabditis elegans. Genetics 92, 419–435. Abstract
*Edited by Victor Ambros. Last revised April 5, 2006. Published June 14, 2006. This chapter should be cited as: Fay, D. Genetic mapping and manipulation: Chapter 6-Mapping with deficiencies and duplications (June 14, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.95.2, http://www.wormbook.org.
Copyright: © 2006 David Fay. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: davidfay@uwyo.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.