Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergUnfortunately for the field of eukaryotic genetics, mutations in most genes do not appear to cause any obvious or penetrant defects (at least under laboratory conditions). Thus, as occurs in the case of C. elegans one is left wondering about the cellular and developmental functions of <70% of the genes in the genome. In some cases, such genes may ultimately be identified in screens for suppressors or enhancers of previously characterized mutations. In addition, our lab and others have developed systematic approaches for identifying synthetic genetic interactions (see Genetic enhancers). The difference between a synthetic mutation and an enhancer mutation is largely semantic. At its most extreme, a synthetic double-mutant strain a b) may exhibit the phenotype C at a very high frequency, whereas neither single mutant ever shows this defect. Alternatively, if both the a and b single mutants display the C phenotype 20% of the time, whereas the a b double mutant exhibits the defect 90% of the time, we would probably refer to a as an enhancer of b or vice versa. Where one draws the line between synthetic mutants and enhancers is arbitrary, although in both cases, the combined effects are always more than additive. Finally, keep in mind that both synthetic and enhancer phenomena are completely allele dependent. In fact, such hidden functions for genes as revealed by synthetic screens may depend heavily on the isolation of rare mutations or weak alleles.
The focus of this section will be on the specific methods our lab has developed to clone bona fide synthetic mutations. Our strategy assumes that the molecular identity of one of the two interacting genes is known and that the presence of the known mutation can be verified by an observable plate phenotype, a molecular lesion, or through a synthetic interaction with another known gene. Some of these approaches will undoubtedly apply to cloning enhancer mutations, particularly when the penetrance of the phenotype in the single mutants is very low. In situations where the penetrance of the phenotype in either single mutant is reasonably high (<25%), however, it may be possible to clone the gene by more straightforward methods. Another significant issue is whether or not double-mutant animals are viable (as is the case for synthetic multivulval mutants). Again, our approach is designed to address the worst-case scenario where the double mutants are not viable and may not even display an easily recognizable plate phenotype. In addition, our method avoids complications resulting from the contribution of maternal products that may significantly reduce the penetrance of the phenotype in the progeny of heterozygous animals. The key to the methods described below is that although labor intensive, unambiguous results can be obtained that will steadily move the mapping process forward. In fact, cloning such synthetic mutations need only be slightly more time consuming than cloning any straightforward mutation.
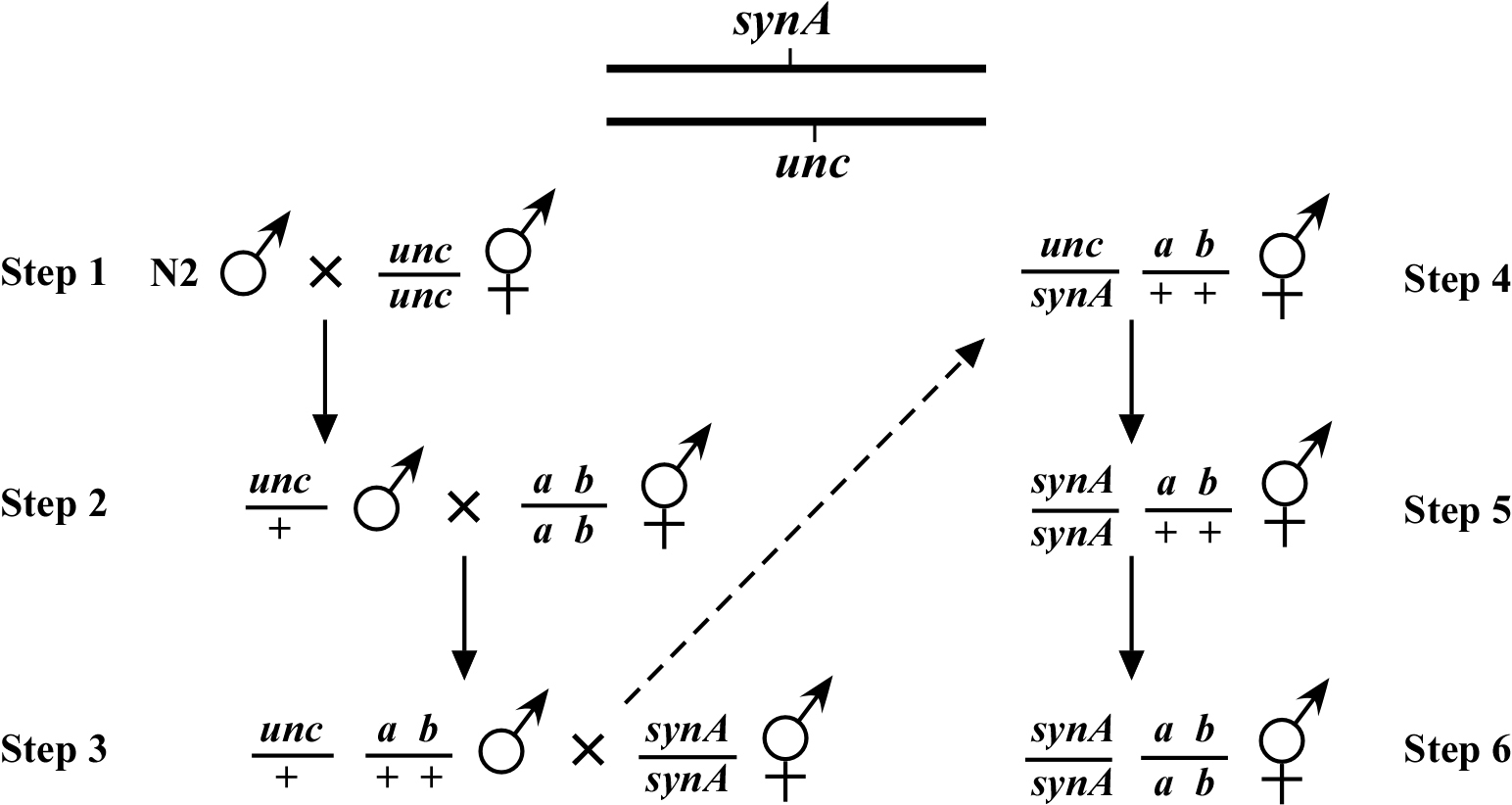
Mapping a synthetic mutation requires building marker strains that contain the known mutation in the background. If the known mutation has a phenotype, then this should be quite straightforward. In situations where the known mutation has no phenotype as a single mutant, however, the question arises: how do you follow its presence or absence? The best way around this is to make use of the opposite chromosome as shown in Figure 1 (Making compound mutants). A visible marker is chosen that maps close to the synthetic mutation synA, in this case an unc. Following mating to N2 males (step 1), the unc/+ (heterozygous) male is next mated to the desired marker strain (step 2) to generate trans-het males, which are then mated into the synA homozygous strain (step 3). We now identify cross-progeny animals that throw both Unc and A B progeny (step 4). By identifying an animal that has lost the unc mutation in the next generation (step 5), we have effectively selected for the synA homozygous mutation. In step 6, the a b mutations are also homozygosed. In the construction of a dpy unc mapping strain, dpy or unc counter markers may be used, although it is necessary that they have phenotypes that are distinguishable from those of the dpy or unc being used as markers.
Although this method requires a fair amount of picking to guarantee selection of the unc/synA; ab/+ animal, it is mostly foolproof, assuming that the counter-marker (in this case unc) is close to the synthetic mutation (synA). Nevertheless, it is wise to generate at least two independent mapping strains to ensure that the correct strain (synA homozygous) is obtained. If known synthetic interactors of synA already exist, these mutations (or RNAi) can be used to test for the presence of synA in the mapping strain. One can also sequence several isolates to verify the presence of the lesion. Alternatively, if the mutation creates a polymorphism or is itself a deletion, PCR methods can be effectively used.
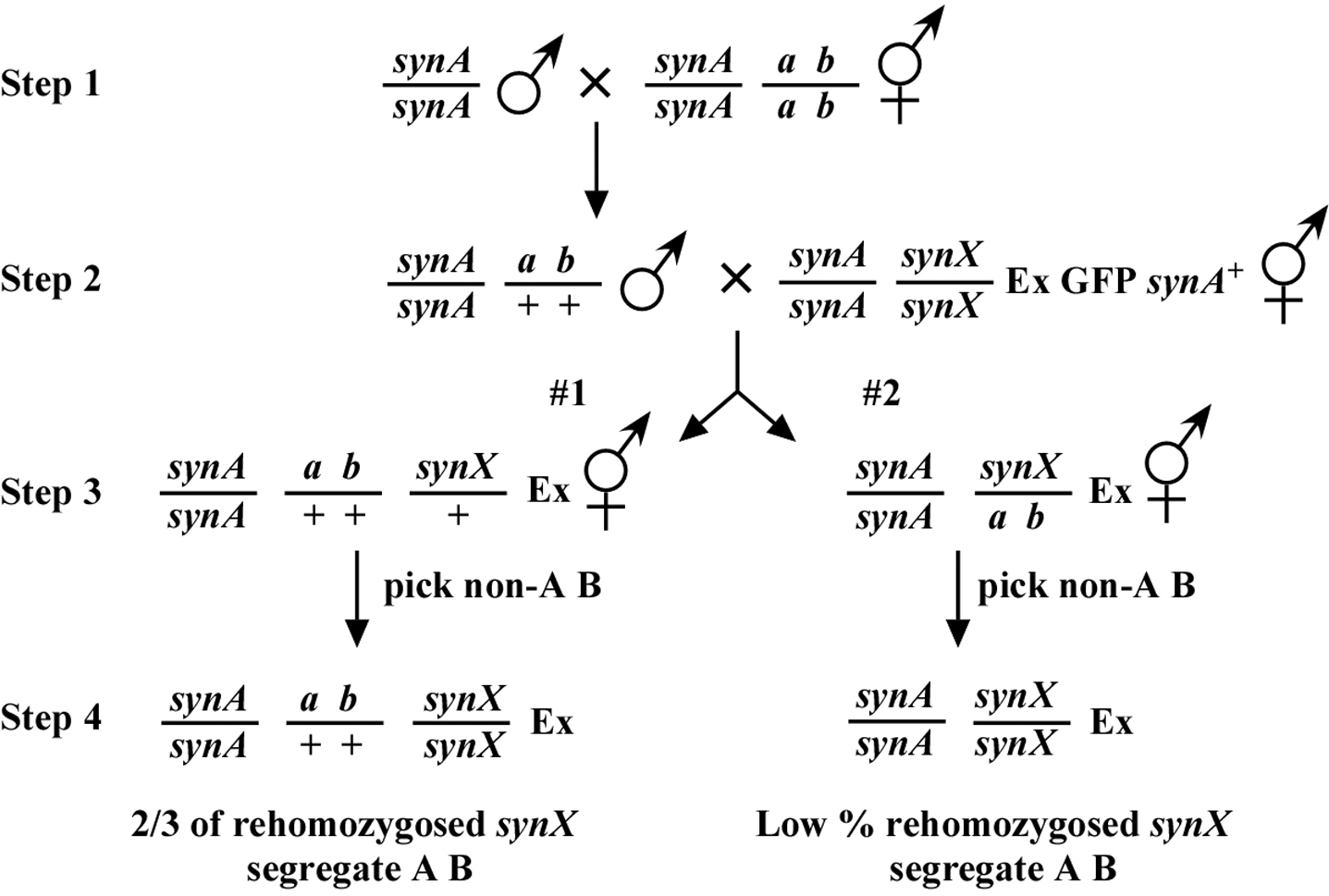
Our described methods for two- and three-point mapping with synthetic mutants require the use of an extrachromosomal array that contains rescuing sequences for the known mutation (syn A) and a widely expressed GFP marker such as sur-5.::GFP. In our case, the arrays we use for mapping are also integral to our isolation of the synthetic mutations. However, such an array could also be derived after the fact. The key is that in homozygous double-mutant strains, only those animals that segregate the rescuing array are viable. Thus, the array allows one to infer the genotype of the animals with respect to the unknown mutation (synX; synA is homozygous throughout). The basic method for two-point mapping is outlined in Figure 2.
In step 1, homozygous synA males are crossed into the mapping strain to generate trans-het males, which are mated to the double-mutant syn strain containing the extrachromosomal array (step 2). Depending on whether or not the synX mutation is on the same chromosome as a b, we have two scenarios. In #1, they are on different chromosomes. Therefore, when we identify progeny where synX is once again homozygous (step 4), 67% of these will throw A B progeny. If syn X is on the same chromosome and close to the markers, rehomozygosed synX animals will fail to throw appreciable A B progeny. All the basic rules of two-point mapping apply here. In this case the frequency of recombinants will be about twice that of the actual map distance (see Two-point mapping for further details; link). Generally speaking, one will want to pick about 100 animals for each chromosome as only 1/4 will be rehomozygosed to score. Note that throughout these steps we will pick only Ex+ animals.
A second source of information comes from the synX non-rehomozygosed plates. When the synX mutation lies on the same chromosome and is close to the markers a b, synX is essentially balanced. Thus nearly all non-rehomozygosed animals for synX will throw A B animals. (In this case the percentage of recombinants will directly equal the map distance.) In contrast, if synX and a b are on separate chromosomes, only 2/3 will throw A B progeny.
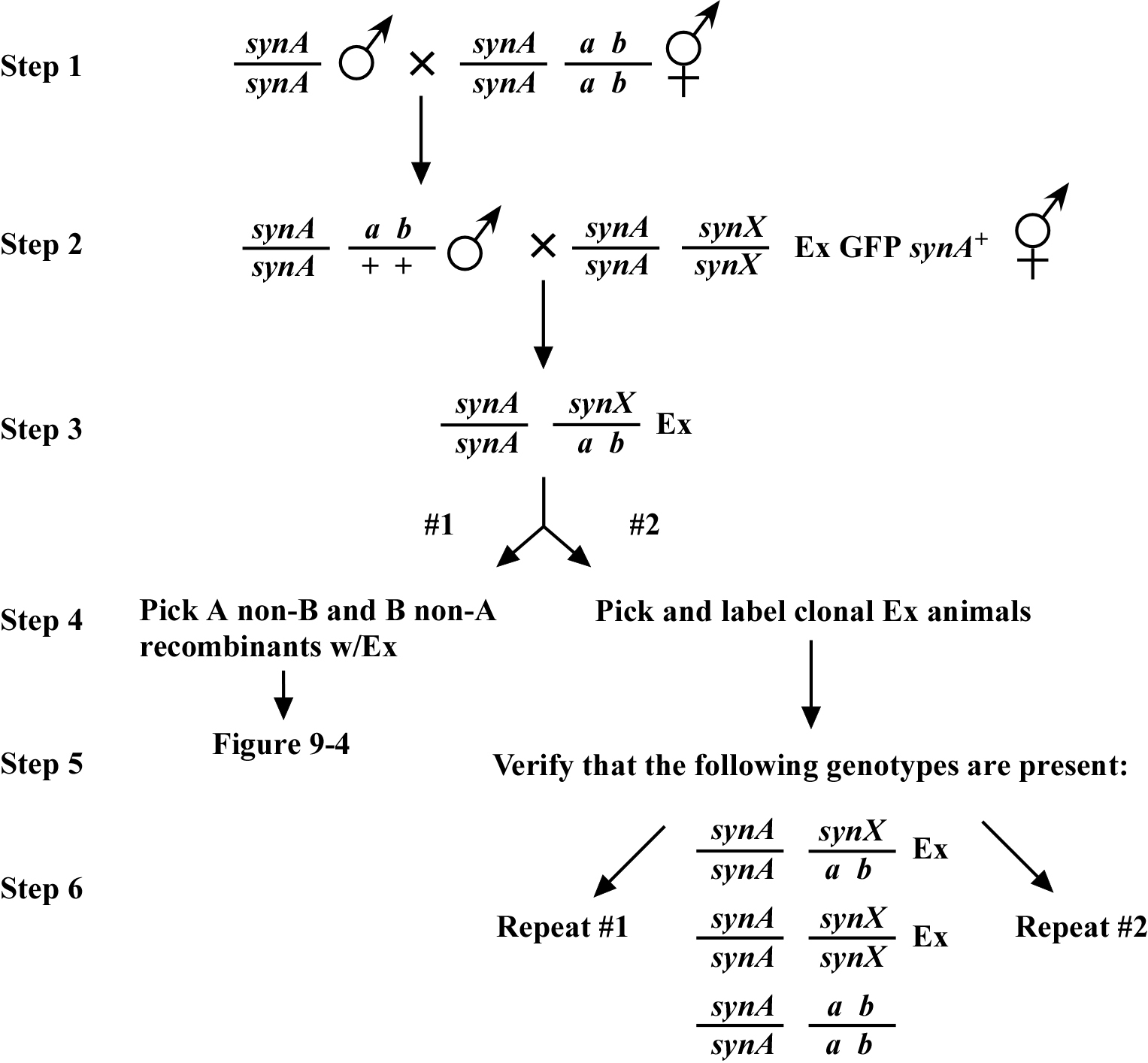
Once assigned to a chromosome, three-point mapping can be undertaken. The approach is reminiscent of the method used for two-point mapping in that we will seek to rehomozygose the synX mutation using the extrachromosomal array after picking recombinants. An important point in mapping a mutation with no phenotype on its own is that we have to be absolutely sure that the synX mutation is actually present in the generation of animals from which we will pick the recombinants. There are essentially two ways to ensure this, as shown in Figure 3.
One is to pick recombinants only immediately after obtaining the trans-heterozygous strain (scheme #1). This guarantees the presence of both synX and the a b marker. This is an effective way to do three-point mapping initially, provided the markers a and b are reasonably far apart (i.e., several map units or more). For markers that are closely spaced, it is often necessary to pick recombinants over several generations or more to get sufficient numbers (scheme #2). Here, however, we run the risk that a recombination event will lead to the loss of the synX mutation, and that this may go undetected. This can happen even though the markers have effectively balanced the silent synX mutation. To ensure that recombinants are picked only from plates where the parent is a true trans-het, we must make certain that ~25% of sibling plates are re-homozygosed. For example, we pick 40 animals (step 4) and 10 turn out to be homozygous for synX (step 5). We then have confidence to pick recombinants off the 30 non-re-homozygosed sibling plates. This strategy can be carried out indefinitely (step 6) until sufficient recombinants are obtained. Note that in picking recombinants as well as propagating the trans-het strain, we will only pick Ex+ animals.
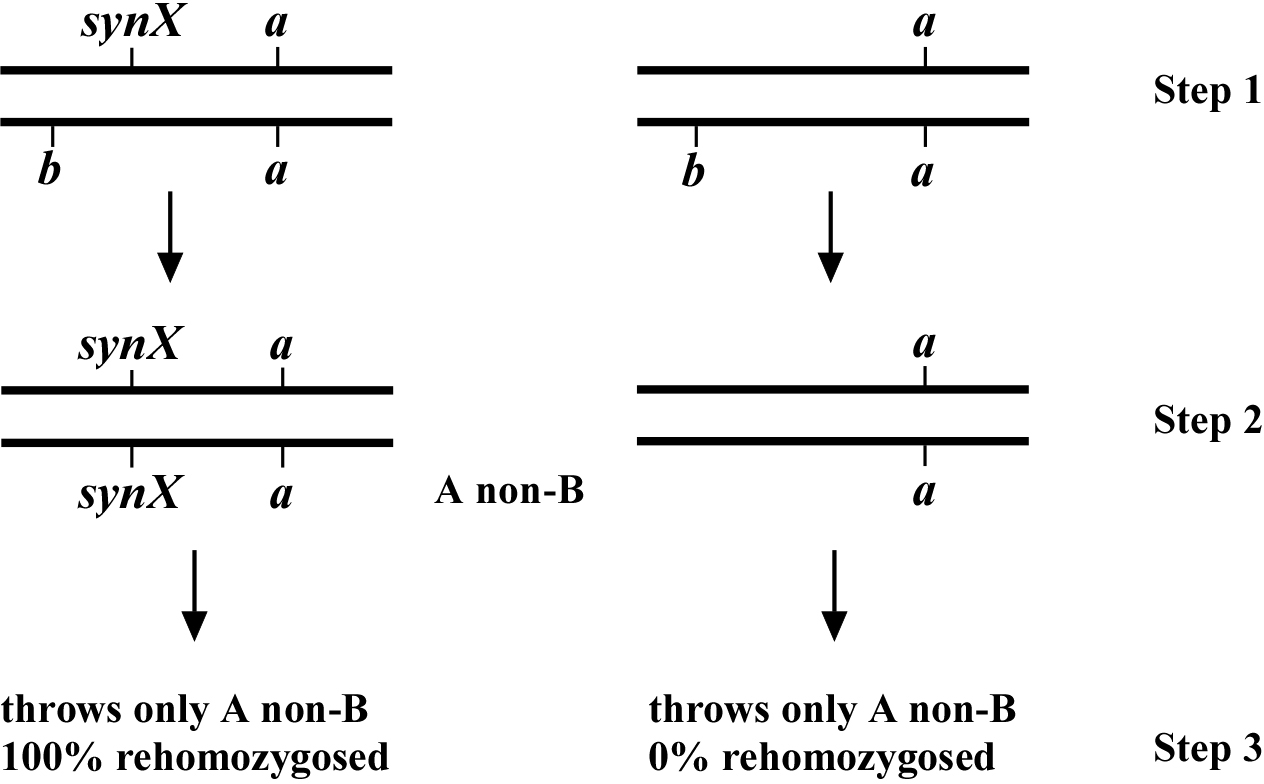
Once a recombinant has been picked, we need to determine whether or not the recombinant chromosome has acquired the synX mutation, as shown in Figure 4. This turns out to be quite straightforward. In the example on the left, an A non-B recombinant has picked up the synX mutation (step 1). This animal will throw additional A non-B progeny, 1/3 of which will be homozygous for the recombinant synX a chromosome (step 2). These animals will require the presence of the rescuing array and can therefore be scored positively for the presence of synX. On the right, synX is not present on the recombinant chromosome and progeny will never throw re-homozygosed synX (step 3). In a typical situation we might pick 15 (Ex +) progeny from an F1 recombinant animal. If the synX mutation has been acquired, 5 animals on average will be homozygous for the recombinant chromosome. For those that are homozygous (based on the absence of a b progeny) we then determine whether these worms also homozygous for synX (i.e, they require the array). Although fairly laborious, this approach will generally give unambiguous data points.
It should be noted that if the mutation lies outside the markers and at some distance, the chance for a second recombination occurring where the syn X mutation is lost in some percentage of the progeny becomes substantial. Thus we might have a situation where five animals are clearly homozygous for the A non-B chromosome, but only four are rehomozygosed with respect to synX. In this case we would count the recombinant as positive for acquiring synX and would also conclude that synX is unlikely to lie between the two markers. This added complexity is not a factor when synX is not initially acquired by the recombinant, as such progeny will never contain the synX mutation.
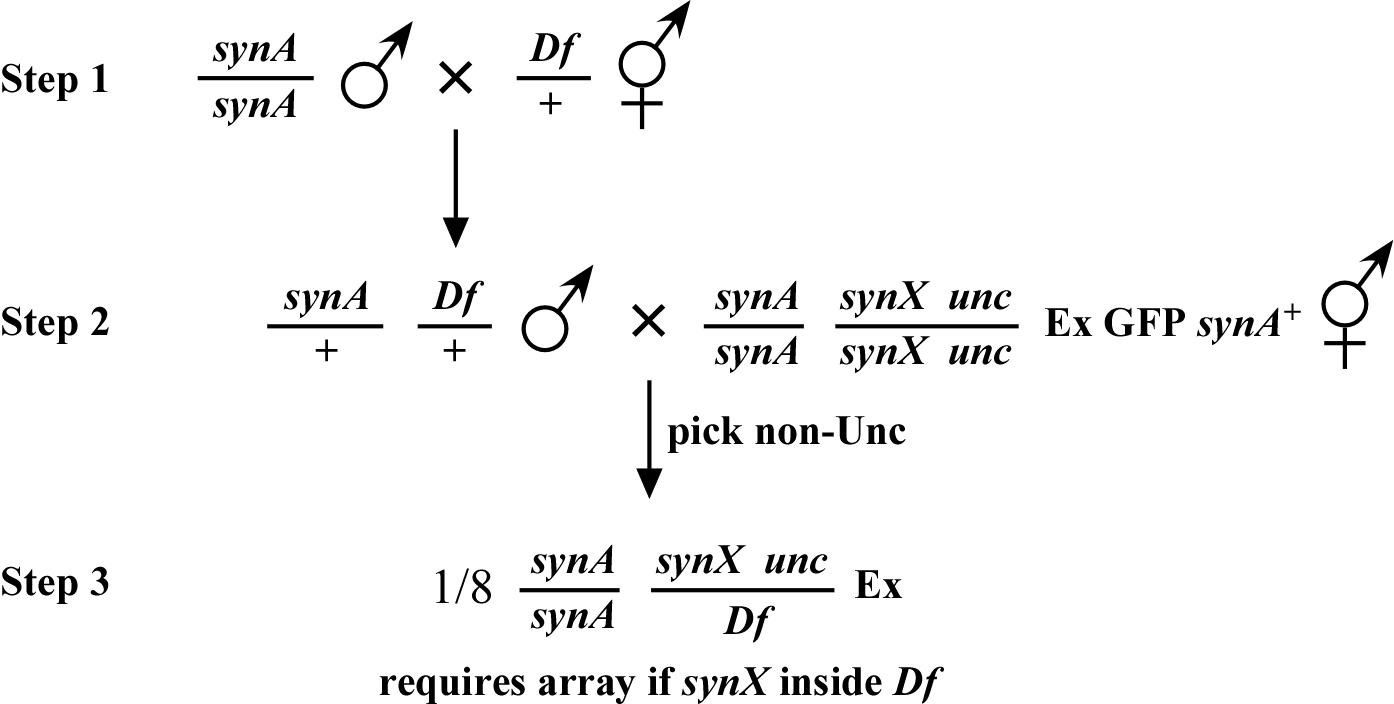
The good news is that we need not construct Df strains that are homozygous for the syn A mutation. The scheme for Df mapping is outlined in Figure 5. Here the Df is crossed into the double-mutant strain containing the array (step 2). The key is to unambiguously determine cross-progeny at this stage. In the example shown above, the chromosome with synX contains a visible marker (unc) in cis, which is outside of the region covered by the deficiency. Thus, the cross-progeny are non-Unc. Alternatively, the double-mutant strain can harbor visible markers on a separate chromosome (such strains are readily obtained during two-point mapping). In the scheme above, 1/8 of the final cross-progeny will be both homozygous for synA and trans-heterozygous for synX and the Df (step 3). Such strains would be identified as non-Uncs that throw Uncs and (most likely) dead eggs and require the array for viability. As discussed in chapter 6, a positive result in deficiency mapping is always much more meaningful than a negative one.
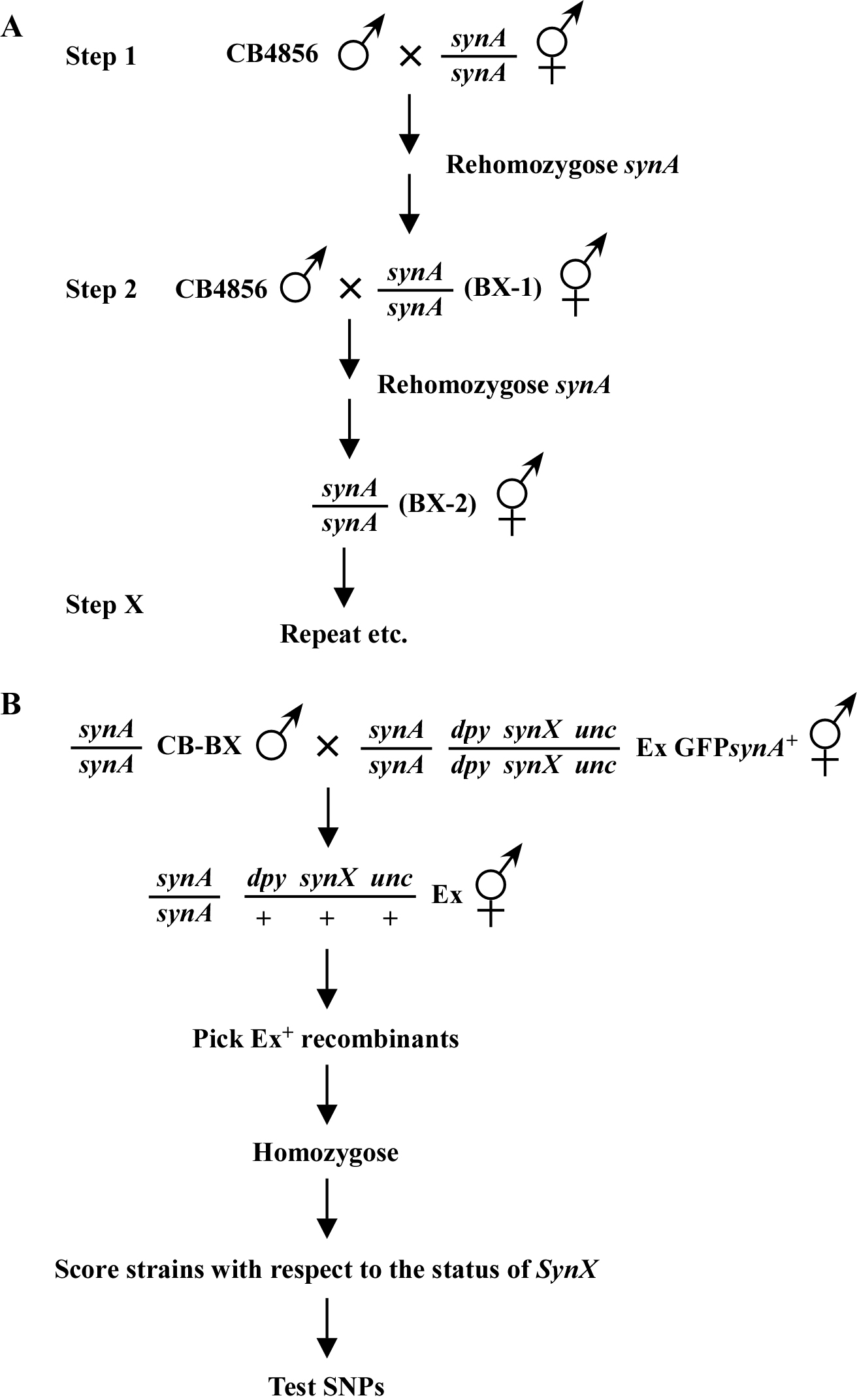
Thankfully, SNPs can be fully employed for both two- and three-point mapping of synthetic mutants. The first critical step is to construct a strain that harbors your mutation of interest in the CB4856 (Hawaiian) background (Figure 6A). This can be a bit tricky, as CB4856 is not sensitive to RNAi feeding and even germline injection of dsRNA is somewhat less potent than it is in N2. The first step is to cross CB4856 males to your known mutation (synA) and then blindly re-isolate candidate homozygous synA mutants. These isolates can then be tested for the presence of homozygous synA by dsRNA injections using a known genetic interactor, PCR-based methods if the mutant in synA causes a RFLP, or direct sequencing. Depending on how many synthetic mutations you anticipate mapping in the future, it may be prudent to successively backcross your mutation to CB4856 to obtain synA strains that have <95% CB4856 DNA content (5 or 6 backcrosses). This is also essential if you are to use SNPs for two-point mapping of your synthetic mutations. Alternatively, if you already have your mutation (synX) mapped to a relatively small region, you could theoretically isolate multiple independent mutant strains after just a single backcross to CB4856. You would then identify one or more isolates that contain CB4856 sequences throughout your region of interest. Of course, all backcrossed strains need to be thoroughly tested to ensure that specific regions are indeed composed exclusively of CB4856 sequence.
Both two-point and three-point mapping methods are largely identical to those already outlined in SNPs: introduction and two-point mapping and SNPs: three-point mapping. Figure 6B shows the basic outline for three-point mapping with SNPs. As for the mapping procedures described above, one always picks Ex+ animals at each step, and the presence of the synX mutation is inferred by the requirement for the array. You may also want to use a two-tiered screening approach as outlined in SNPs: three-point mapping to maximize your efficiency.
*Edited by Victor Ambros. Last revised May 7, 2005. Published February 17, 2006. This chapter should be cited as: Fay, D. Genetic mapping and manipulation: Chapter 9-Synthetic and enhancer mutations (February 17, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.98.1, http://www.wormbook.org.
Copyright: © 2006 David Fay. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: davidfay@uwyo.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.